Novel heterozygous FOXN1 mutation identified following newborn screening for severe combined immunodeficiency is associated with improving immune parameters
Abstract
Background: Forkhead-box protein N1 (FOXN1) plays a critical role in the proper development and function of thymic epithelial cells, required for T cell ontogeny. Homozygous variants in the FOXN1 gene, encoding FOXN1, cause severe combined immunodeficiency (SCID), whereas heterozygous mutations are associated with variable presentations and over time, improving T cell function.
Aim: To highlight the importance of broader genetic investigations to attain a definitive molecular diagnosis following abnormal newborn screening for SCID.
Methods: Case report of a patient with immunodeficiency due to a novel de novo FOXN1 mutation.
Results: The patient was identified following abnormal newborn screening for SCID in which T cell receptor excision circles were absent/very low. Initial immune investigations revealed severe T cell lymphopenia and poor lymphocyte function and she was diagnosed with T-B+NK+SCID. During work-up for hematopoietic stem cell transplantation, extensive genetic investigations identified a novel heterozygous mutation in FOXN1. A more conservative management approach was taken, and over the following months, the patient’s immune parameters improved.
Conclusion: Newborn screening for SCID has facilitated the detection of SCID, as well as other T cell immunodeficiencies, before infectious complications and organ damage occur. Heterozygous mutations in FOXN1 are associated with more variable presentations including improving immune indices with age. Here, results of genetic investigations were essential for informing the management of this case.
Statement of Novelty: We report a novel heterozygous mutation in FOXN1, presenting initially as T-B+NK+ SCID with gradual improvement of immune parameters over time.
Introduction
The Forkhead-box (FOX) superfamily of transcription factors play important roles in the homeostasis, function, and aging of a variety of organs and tissues – including the immune system. FOX protein N1 (FOXN1), encoded by the FOXN1 gene, is an essential regulator of thymic epithelial cell differentiation required for T cell development, and is also expressed in skin and hair follicles (Vaidya et al. 2016). Potential roles in hematopoiesis within the bone marrow have been reported (Zook et al. 2013).
The thymus provides a unique environment for the differentiation, selection, and maturation of bone marrow-derived T cell progenitors into fully functional T cells. This process relies on the ordered architecture of the thymus, consisting of a cortical area rich in cortical thymic epithelial cells (cTECs) and lymphoid cells, a medullary area containing medullary thymic epithelial cells (mTECs), Hassall’s corpuscles, macrophages, dendritic cells and B lymphocytes, as well as a transitional corticomedullary junction. Importantly, activation of FOXN1 and subsequent transcriptional regulation of downstream genes are needed for the proper differentiation, maturation, and function of cTECs and mTECs during prenatal thymus organogenesis, as well as homeostasis of the postnatal thymus (Corbeaux et al. 2010; Vaidya et al. 2016). Thus, loss-of-function of this so-called master regulator results in ablated thymic and T cell development, and the phenotype of severe combined immunodeficiency (SCID).
Bi-directional cues between the thymic stroma and T cells are essential for the normal development and function of both entities (van Ewijk et al. 1994). Defective thymic architecture has been documented in instances where T cells exhibit stunted development (Palmer et al. 1993), whereas reconstitution of the thymic microenvironment has been reported with the administration of T cell-committed precursors (Roberts et al. 2009). Importantly, animal models have been an essential tool for dissecting the role of FOXN1 during early thymus development (Bosticardo et al. 2019). Mice deficient in Foxn1 (‘nude’ spontaneous mutation) are athymic, lack T cells, and exhibit abnormal hair growth (Nehls et al. 1994). Among the panel of genes regulated by foxn1 are Ccl25, required for homing of immature thymocytes to the thymus (Liu and Leung, 2006), Dll4, involved in the Notch pathway-directed sorting of cells towards T cell lineage (Koch et al. 2008), and Cxcl12, needed for expansion of T cell precursors in the thymus (Ara et al. 2003). Other genes induced by foxn1 required for thymic epithelial cell and thymocyte exchange are Psmb11, Prss16, and Cd83 (Bosticardo et al. 2019).
In humans, null mutations in FOXN1 are characterized by severe T cell immunodeficiency (T-B+NK+ phenotype), congenital alopecia, and nail dystrophy (OMIM #601705) (Amorosi et al. 2008; Frank et al. 1999; Pignata et al. 1996). Affected individuals suffer recurrent infections, failure to thrive, and oral candidiasis. While some attempts to correct the deficiency have been made with hematopoietic stem cell transplantation (HSCT), overall, post-transplant outcomes for individuals with defects in FOXN1 indicate that thymus transplants are a more effective and curative option (Bosticardo et al. 2019; Chou et al. 2014).
Monoallelic mutations in FOXN1 are more frequently associated with variable clinical features. A recent longitudinal analysis of 25 children and 22 adults with heterozygous FOXN1 variants described the majority being clinically well except for viral respiratory infections (Bosticardo et al. 2019). Common non-infectious manifestations included eczema and nail dystrophy. Of the children, 21 were identified following an abnormal newborn screen (NBS) result and were associated with T cell lymphopenia. For most of these cases, CD4+ T cells eventually recovered.
Here, we report on our experience with an infant who was diagnosed with T-B+NK+ severe combined immunodeficiency (SCID) following an abnormal NBS result. While undergoing work up for HSCT, extensive genetic investigations including whole exome sequencing (WES) and whole genome sequencing (WGS) identified a novel heterozygous variant in FOXN1, which subsequently changed the management plan for this patient.
Case presentation
The patient is currently a 19-month-old female, the first child to non-consanguineous parents of Asian descent. The pregnancy was uncomplicated and she was born at 39+2 weeks gestation by spontaneous vaginal delivery. Her mother had 2 prior early miscarriages and her father has 2 healthy older children from a previous marriage. The patient came to the attention of the Immunology team following an abnormal NBS result for SCID. The initial T cell receptor excision circle (TREC) level was 0 (cut-off: 75), and the repeat TREC was 7. Immunodeficiencies associated with ADA, PNP, ZAP70, and IKBKB were ruled out during initial screening tests, and assessment for microdeletions in 22q11.6 was returned negative.
Initial Evaluations
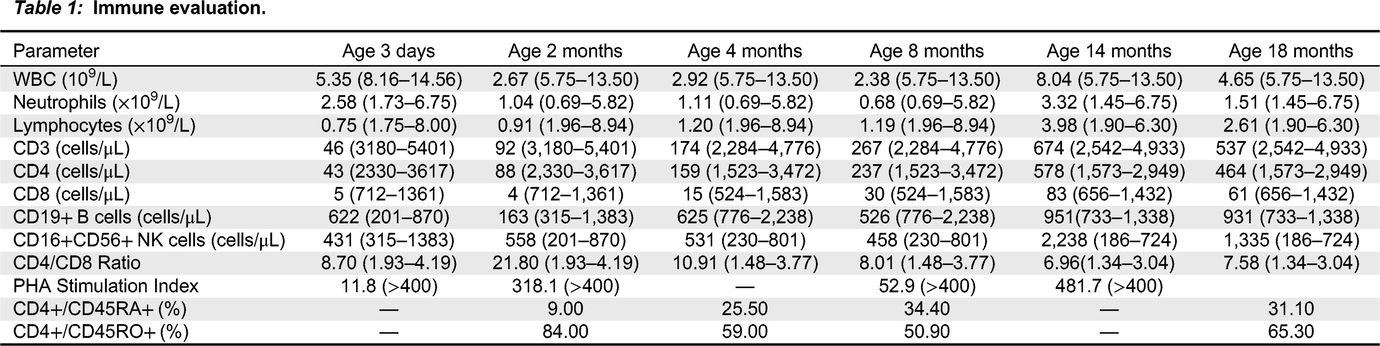
Initial immune evaluation revealed T cell lymphopenia of both CD4+ cells (43 cells/μL, normal: 2330-3617 cells/μL) and CD8+ cells (5 cells/μL, normal: 712-1361 cells/μL). B cells (431 cells/μL, normal: 315-1383 cells/μL) and NK cells (622 cells/μL, normal: 201-870 cells/μL) were within normal range. Output of naïve T cells was low at 9% (CD4+/CD45RA+), while memory T cells predominated at 84% (CD4+/CD45RO+). Recent thymic emigrants were low at 9% (normal: 64–94%). Her lymphocyte proliferation responses to the mitogen phytohemagglutinin (PHA) were very low at 12 (stimulation index, SI normal >400) (Table 1). Ultrasound of the neck revealed normal size lymph nodes in both jugular chains; however, there was no convincing evidence of thymic tissue.
Table 1:
Genetics
Genetic work-up to identify a causative molecular defect underlying the patient’s immunodeficiency was started. An initial SCID gene panel (Newborn Screening Ontario; 20 genes) was negative. Subsequently, a more extensive PID panel (Blueprint Genetics) was sent which identified, at 2 months of age, a novel heterozygous frameshift mutation in FOXN1 (NM_003593.2), c.1465del, resulting in p. Gln489Argfs*61. This variant was absent from large population databases (gnomAD). To ensure that no other genetic aberrations were present, trio WGS of the patient and her parents were sent. While no further changes were found (including non-coding or copy number variants), the mutation was identified as a de novo variant targeting exon 7 — a region where many pathogenic variants reside.
Management
Our patient’s initial presentation was in keeping with T-B+NK+ SCID, and she was managed accordingly with antibiotic prophylaxis and immunoglobulin replacement therapy (from 4 weeks of age) while workup for HSCT was initiated. After the pathogenic mutation in FOXN1 was identified, in line with our center’s experience with patients harboring heterozygous variants in FOXN1, we instead changed our approach to ‘watchful waiting’ to see if her immune parameters would improve.
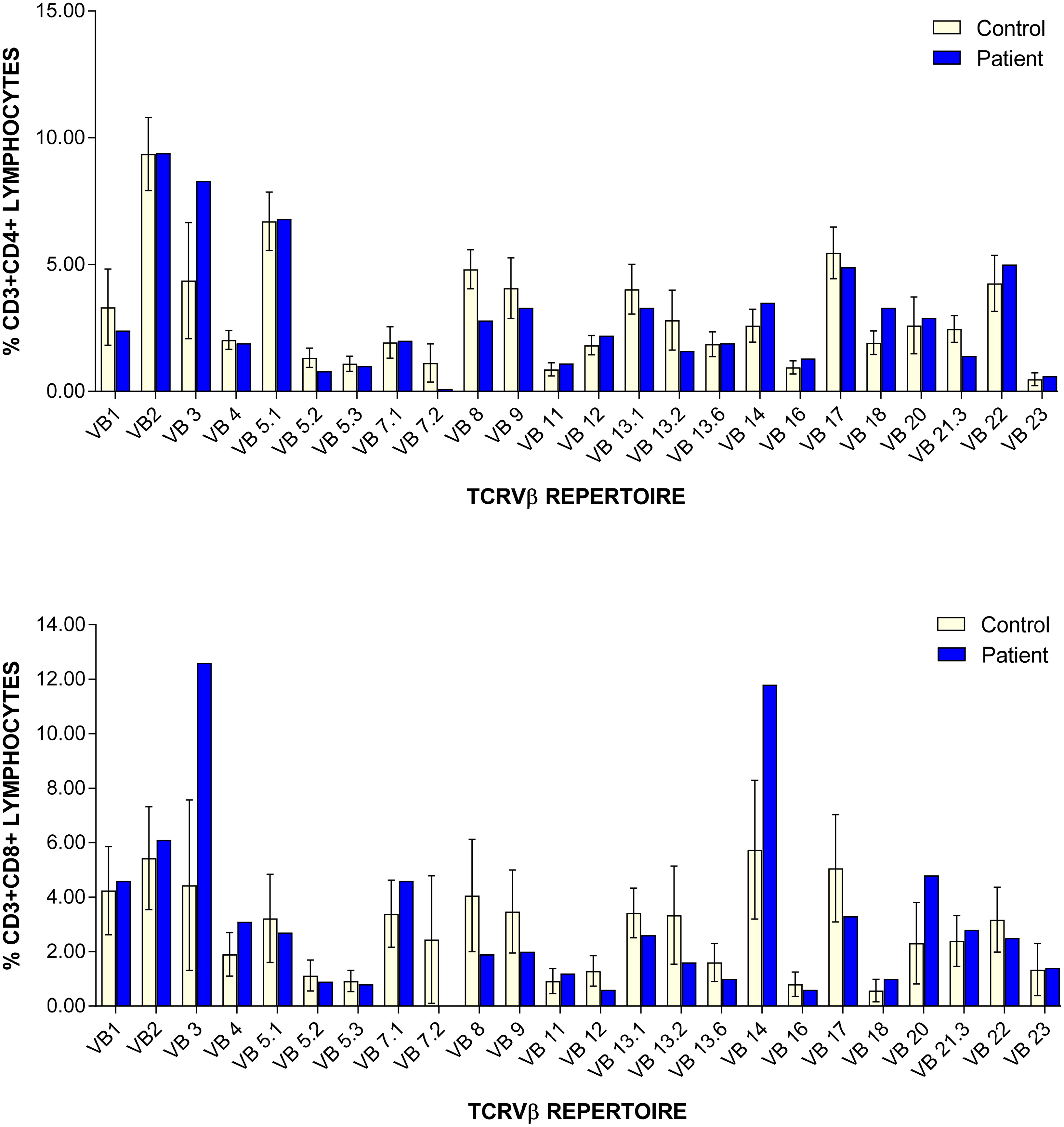
In the subsequent months, the patient’s lymphocyte function recovered (PHA stimulation index increased gradually to 481.7), and her T cell counts continue to show improvement although are still low (Table 1). CD45RA/RO assessment revealed improving naïve T cells (31.10%) versus memory T cells (65.30%). Recent thymic emigrants are steadily increasing 21% (normal: 40–100%). Assessment of TCRVβ repertoire revealed only slight under-representation of CD4+ Vβ8 and Vβ21.3 clones, and over-representation of CD8+ Vβ3 and Vβ14 clones (Figure 1). At 8 months of age, antibody replacement therapy was stopped and her immunoglobulin levels are currently within normal range (IgG: 5.1 g/L (normal: 3.2–11.5 g/L); IgA: 0.2 g/L (normal: 0–0.9 g/L); IgM: 0.5 g/L (normal: 0.5–1.9 g/L)). She has received her routine non-live vaccines and is responding well (anti-tetanus toxoid IgG >5 IU/mL).
Figure 1:
To date, the patient remains clinically well and has not had any significant infections. There is presently no indication for HSCT and she continues to be followed closely.
Discussion
The implementation of SCID NBS has transformed our ability to identify and treat infants with SCID before infectious complications and end-organ damage occurs (Kwan et al. 2014). Screening utilizes measurement of TRECs, which are a by-product of T cell receptor recombination. Thus, very low/absent TRECs in neonates are an indication of defects in T cell development and prompts the diagnostic evaluation for SCID (Biggs et al. 2017). By extension, less profound immunodeficiencies that would not have been detected by past practices until individuals presented clinically are also being flagged for follow-up (Amatuni et al. 2019; Mandola et al. 2019; Scott et al. 2021).
Our patient was initially diagnosed with T-B+NK+ SCID following abnormal SCID NBS with two absent/low TREC determinations. While she underwent workup for HSCT, extensive genetic investigations identified a single novel de novo heterozygous mutation in FOXN1. Our center’s experience with heterozygous FOXN1 mutations (Scott et al. 2021) as well as those reported in the literature, changed the intended management plan to a more conservative stance given the likelihood that the patient’s immune parameters would improve with age. Moreover, HSCT is unlikely to correct the lymphopenia associated with a poorly developed thymus (Bosticardo et al. 2019; Chou et al. 2014). Our case is unique in that the genetic work-up involved gene panel sequencing, WES, and WGS to rule out the possibility of another underlying genetic etiology, affirming our approach to closely monitor the patient for improvement.
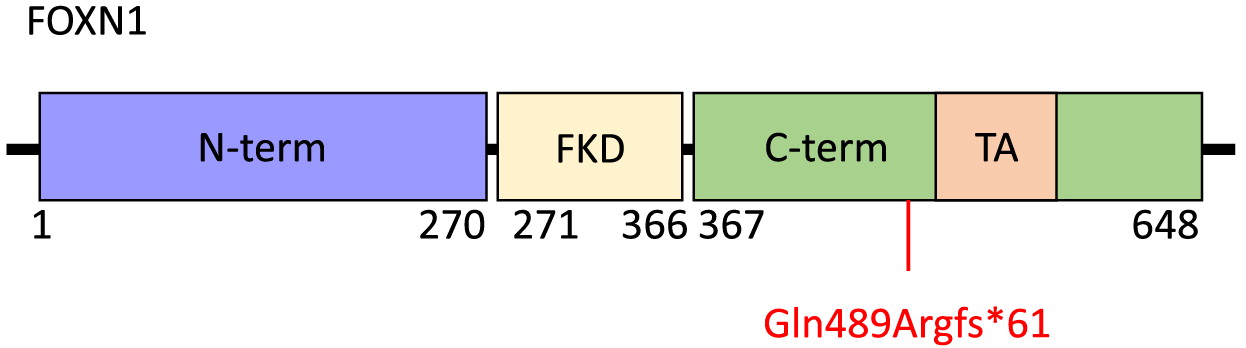
FOXN1 is a 648-amino acid protein with an N-terminal region of residues 1–270, a Forkhead domain located centrally between amino acids 271 and 366, and a C-terminal region comprising residues 367–648 (Figure 2). Within the C-terminal region is an evolutionarily conserved transcriptional activation domain. The N-terminal region has been implicated in thymic epithelial cell differentiation while the C-terminal region is required for high-affinity DNA binding (Newman et al. 2020). The FOXN1 frameshift mutation identified in our patient, c.1465del (p.Gln489Argfs*61), targets exon 7 and causes a premature stop at position 61 within the C-terminal domain. This is predicted to result in loss of normal protein function due to truncation of the protein or nonsense-mediated mRNA decay. The clinical picture of our patient, with CD4+ and CD8+ T cell lymphopenia and unaffected B and NK cell counts is not dissimilar to other reports of patients with mutations within the C-terminal domain, nor of other domains within FOXN1 (Bosticardo et al. 2019). Thus, it remains to be seen whether a genotype-phenotype correlation exists.
Figure 2:
Athymia has been reported in individuals with pathogenic homozygous FOXN1 mutations (Chen et al. 2009) as well as heterozygous variants (Bosticardo et al. 2019). Of the 13 infants reported by Bosticardo and colleagues, only 3 had a normal-sized thymic shadow while the remaining were absent or reduced in size. In our patient, ultrasound did not detect evidence of thymic tissue. Nevertheless, the presence of near normal TCRV beta repertoire indicates that the thymus is likely small or perhaps ectopically placed (Shah et al. 2001).
In summary, this report highlights the importance of robust genetic investigations to delineate the underlying cause of immunodeficiencies detected by SCID NBS. We continue to follow a conservative approach with this patient, particularly as more reports of disease course and outcomes of individuals affected by heterozygous FOXN1 mutations come to light.
REFERENCES
Acuna-Hidalgo R., Veltman J.A., and Hoischen A. 2016. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17(1): 241.
Amatuni G.S., Currier R.J., Church J.A., Bishop T., Grimbacher E., Nguyen A.A.-C., Agarwal-Hashmi R., Aznar C.P., Butte M.J., Cowan M.J., Dorsey M.J., Dvorak C.C., Kapoor N., Kohn D.B., Markert M.L., Moore T.B., Naides S.J., Sciortino S., Feuchtbaum L., and Puck J.M. 2019. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California, 2010–2017. Pediatrics, 143(2): e20182300.
Amorosi S., D’Armiento M., Calcagno G., Russo I., Adriani M., Christiano A.M., Weiner L., Brissette J.L., and Pignata C. 2008. Foxn1 homozygous mutation associated with anencephaly and severe neural tube defect in human athymic nude/SCID fetus. Clinical Genetics, 73(4): 380–384.
Ara T., Itoi M., Kawabata K., Egawa T., Tokoyoda K., Sugiyama T., Fujii N., Amagai T., and Nagasawa T. 2003. A role of CXC chemokine ligand 12/stromal cell-derived factor-1/pre-B cell growth stimulating factor and its receptor CXCR4 in fetal and adult T cell development in vivo. J. Immunol. 170(9): 4649–4655.
Biggs C.M., Haddad E., Issekutz T.B., Roifman C.M., and Turvey S.E. 2017. Newborn screening for severe combined immunodeficiency: A Primer for clinicians. Can. Med. Assoc. J. 189(50).
Bosticardo M., Yamazaki Y., Cowan J., Giardino G., Corsino C., Scalia G., Prencipe R., Ruffner M., Hill D.A., Sakovich I., Yemialyanava I., Tam J.S., Padem N., Elder M.E., Sleasman J.W., Perez E., Niebur H., Seroogy C.M., Sharapova S., and Notarangelo L.D. 2019. Heterozygous FOXN1 variants cause low trecs and severe T cell lymphopenia, revealing a crucial role of Foxn1 in supporting early thymopoiesis. Am. J. Hum. Genet. 105(3): 549–561.
Chen L., Xiao S., and Manley N.R. 2009. FOXN1 is required to maintain the postnatal thymic microenvironment in a dosage-sensitive manner. Blood, 113(3): 567–574.
Chou J., Massaad M.J., Wakim R.H., Bainter W., Dbaibo G., and Geha R.S. 2014. A novel mutation in FOXN1 resulting in SCID: A case report and literature review. Clinical Immunol. 155(1): 30–32.
Corbeaux T., Hess I., Swann J.B., Kanzler B., Haas-Assenbaum A., and Boehm T. 2010. Thymopoiesis in mice depends on a foxn1-positive thymic epithelial cell lineage. Proc. National Academy Sci. 107(38): 16613–16618.
Cowan J.E., Takahama Y., Bhandoola A., and Ohigashi I. 2020. Postnatal involution and counter-involution of the thymus. Front Immunol. 11: 897.
Currier R. and Puck J.M. 2021. SCID newborn screening: What we’ve learned. J. Allergy Clinical Immunol. 147(2): 417–426.
Du Q., Huynh L.K., Coskun F., Molina E., King M.A., Raj P., Khan S., Dozmorov I., Seroogy C.M., Wysocki C.A., Padron G.T., Yates T.R., Markert M.L., de la Morena M.T., and van Oers N.S.C. 2019. FOXN1 compound heterozygous mutations cause selective thymic hypoplasia in humans. J. Clin. Invest. 129(11): 4724–4738.
Frank J., Pignata C., Panteleyev A.A., Prowse D.M., Baden H., Weiner L., Gaetaniello L., Ahmad W., Pozzi N., Cserhalmi-Friedman P.B., Aita V.M., Uyttendaele H., Gordon D., Ott J., Brissette J.L., and Christiano A.M. 1999. Exposing the human nude phenotype. Nature, 398(6727): 473–474.
Koch U., Fiorini E., Benedito R., Besseyrias V., Schuster-Gossler K., Pierres M., Manley N.R., Duarte A., MacDonald H.R., and Radtke F. 2008. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J. Exp. Med. 205(11): 2515–2523.
Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., Baker M., Ballow M., Bartoshesky L.E., Bonilla F.A., Brokopp C., Brooks E., Caggana M., Celestin J., Church J.A., Comeau A.M., Connelly J.A., Cowan M.J., Cunningham-Rundles C., and Puck J.M. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA, 312(7): 729.
Liu H. and Leung B.P. 2006. CD4+CD25+ regulatory T cells in health and disease. Clin. Exp. Pharmacol. Physiol. 33(5–6): 519–524.
Mandola A.B., Reid B., Sirror R., Brager R., Dent P., Chakroborty P., Bulman D.E., and Roifman C.M. 2019. Ataxia telangiectasia diagnosed on newborn screening–case cohort of 5 Years’ experience. Front Immunol. 10: 2940.
Nehls M., Pfeifer D., Schorpp M., Hedrich H., and Boehm T. 1994. New member of the winged-helix protein family disrupted in mouse and rat nude mutations. Nature, 372(6501): 103–107.
Newman J.A., Aitkenhead H., Gavard A.E., Rota I.A., Handel A.E., Hollander G.A., and Gileadi O. 2020. The crystal structure of human forkhead box N1 in complex with DNA reveals the structural basis for forkhead box family specificity. J. Biol. Chem. 295(10): 2948–2958.
Palmer D.B., Viney J.L., Ritter M.A., Hayday A.C., and Owen M.J. 1993. Expression of the αβ T-cell receptor is necessary for the generation of the thymic medulla. Dev. Immunol. 3(3): 175–179.
Pignata C., Fiore M., Guzzetta V., Castaldo A., Sebastio G., Porta F., and Guarino A. 1996. Congenital alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. Am. J. Med. Genet. 65(2): 167–170.
Radha Rama Devi A., Panday N.N., and Naushad S.M. 2017. Foxn1 Italian founder mutation in Indian family: Implications in prenatal diagnosis. Gene. 627: 222–225.
Roberts N.A., Desanti G.E., Withers D.R., Scott H.R., Jenkinson W.E., Lane P.J., Jenkinson E.J., and Anderson G. 2009. Absence of thymus crosstalk in the fetus does not preclude hematopoietic induction of a functional thymus in the adult. European J. Immunol. 39(9): 2395–2402.
Rode I., Martins V.C., Küblbeck G., Maltry N., Tessmer C., and Rodewald H.-R. 2015. FOXN1 protein expression in the developing, aging, and regenerating thymus. J. Immunol. 195(12): 5678–5687.
Romano R., Palamaro L., Fusco A., Giardino G., Gallo V., Del Vecchio L., and Pignata C. 2013. Foxn1: A master regulator gene of thymic epithelial development program. Front Immunol. 4: 187.
Rota I.A. and Dhalla F. 2017. FOXN1 deficient nude severe combined immunodeficiency. Orphanet J. Rare Dis. 12(1).
Rota I.A., Handel A.E., Maio S., Klein F., Dhalla F., Deadman M.E., Cheuk S., Newman J.A., Michaels Y.S., Zuklys S., Prevot N., Hublitz P., Charles P.D., Gkazi A.S., Adamopoulou E., Qasim W., Davies E.G., Hanson I., Pagnamenta A.T., and Holländer G.A. 2021. Foxn1 forms higher-order nuclear condensates displaced by mutations causing immunodeficiency. Sci. Adv. 7(49): eabj9247.
Ruan L., Zhang Z., Mu L., Burnley P., Wang L., Coder B., Zhuge Q., and Su D.-M. 2014. Biological significance of FoxN1 gain-of-function mutations during T and B lymphopoiesis in juvenile mice. Cell Death Dis. 5(10): e1457.
Scott O., Garkaby J., Willett-Pachul J., Mandola A.B., and Pasternak Y. 2021. A novel splice site variant in FOXN1 in a patient with abnormal newborn screening for severe combined immunodeficiency and congenital lymphopenia. LymphoSign J. 8(1): 1–4.
Shah S.S., Lai S.Y., Ruchelli E., Kazahaya K., and Mahboubi S. 2001. Retropharyngeal aberrant thymus. Pediatrics, 108(5): E94.
Vaidya H.J., Briones Leon A., and Blackburn C.C. 2016. Foxn1 in thymus organogenesis and development. Europ. J. Immunol. 46(8): 1826–1837.
van Ewijk W., Shores E.W., and Singer A. 1994. Crosstalk in the mouse thymus. Immunol. Today, 15(5): 214–217.
Zook E.C., Zhang S., Gerstein R.M., Witte P.L., and Le P.T. 2013. Enhancing T lineage production in aged mice: A novel function of Foxn1 in the bone marrow niche. J. Immunol. 191(11): 5583–5593.
Žuklys S., Handel A., Zhanybekova S., Govani F., Keller M., Maio S., Mayer C.E., Teh H.Y., Hafen K., Gallone G., Barthlott T., Ponting C.P., and Holländer G.A. 2016. Foxn1 regulates key target genes essential for T cell development in postnatal thymic epithelial cells. Nat. Immunol. 17(10): 1206–1215.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 2 • June 2022
Pages: 45 - 51
History
Received: 24 May 2022
Accepted: 30 May 2022
Accepted manuscript online: 30 May 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Laura EdithAbrego Fuentes, JennyGarkaby, JessicaWillett Pachul, AbbyWatts-Dickens, MeghanFraser, Vy H.D.Kim, and Chaim M.Roifman. 2022. Novel heterozygous FOXN1 mutation identified following newborn screening for severe combined immunodeficiency is associated with improving immune parameters. LymphoSign Journal.
9(2): 45-51. https://doi.org/10.14785/lymphosign-2022-0007
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
