CTLA4 haploinsufficiency caused by a novel heterozygous splice site mutation
Abstract
Background: Cytotoxic T lymphocyte-associated antigen-4 (CTLA4) haploinsufficiency is characterized by a variety of phenotypes, ranging from autoimmune disorders, enteropathy, fatal combined immunodeficiency, as well as lymphoproliferation and malignancy.
Aim: To broaden the genotypic spectrum and clinical presentations of patients with CTLA4 variants.
Methods: We evaluated a female patient with autoimmunity and lymphopenia. Immune workup and whole exome sequencing (WES) were performed.
Results: The proband presented at 11 years of age with hypothyroidism and later developed Evans syndrome, alopecia, eczema, and lymphocytic interstitial pneumonia. Immune evaluation revealed T, B, and NK lymphopenia with normal humoral immunity. Following a negative genetic panel for autoimmune lymphoproliferative syndrome (ALPS), WES analysis identified a novel heterozygous intronic variant predicted in-silico to cause skipping of exon 2 of the CTLA4 gene.
Conclusion: A novel heterozygous mutation in CTLA4 caused variable presentations of immune dysregulation, one of the hallmarks of CTLA4 haploinsufficiency.
Statement of Novelty: We herein report a novel mutation in CTLA4 resulting in various features of autoimmunity.
Background
Cytotoxic T lymphocyte-associated antigen-4 (CTLA4) is a transmembrane receptor that acts as a checkpoint to dampen T cell-mediated responses and maintain immune self-tolerance. It serves as the opposing face of the CD28 signaling pathway, the latter of which drives T cell activation, differentiation and effector responses, as well as T follicular helper cell and regulatory T cell (Treg) generation and survival. CTLA4 is localized to the plasma membrane and various intracellular compartments in Tregs, and can be upregulated by conventional T cells. Binding of CTLA4 with the shared CD28 costimulatory receptor ligands, CD80 and CD86, found on antigen presenting cells, leads to ligand internalization/degradation and subsequent impairment of archetypal CD28-dependent signaling and T cell responses. Importantly, the ability to capture these ligands is dependent on sufficient CTLA4 cell surface expression. Defects in CTLA4 expression or internalization pathways result in immune dysregulation (Tivol et al. 1995; Lo et al. 2015).
The critical role of CTLA4 in maintaining immune tolerance is supported by findings in murine models showing fatal autoimmunity, including lymphoproliferation and multiorgan tissue damage, within the first few weeks of life (Tivol et al. 1995; Waterhouse et al. 1995). In humans, heterozygous (autosomal dominant) pathogenic mutations in the CTLA4 gene were shown to cause quantitative reductions in CTLA4 expression, leading to loss of peripheral tolerance, infiltrative autoimmune disease, enteropathy, immune dysregulation, as well as immunodeficiency and malignancy such as lymphoma and gastric cancer (Kuehn et al. 2014; Schubert et al. 2014; Hayakawa et al. 2016; Schwab et al. 2018). Interestingly, the presentation of CTLA4 deficiency varies widely, even within kindreds bearing the same mutation. Schubert et al (2014) described a family with 11 members carrying the same heterozygous mutation in CTLA4; 5 suffered from hypogammaglobulinemia, autoimmune cytopenia, recurrent respiratory infections and lung disease, while the other 6 remained asymptomatic.
Treatment of CTLA4 haploinsufficiency includes immunosuppressants, immunomodulatory drugs such as Rapamycin, biologics including Rituximab, immunoglobulin replacement therapy, splenectomy, and hematopoietic stem cell transplantation (HSCT) in rare cases. Therapy is mostly based on clinicians’ judgement and takes into consideration specific organ involvement and immunological assessment (Schwab et al. 2018). Importantly, treatment with Abatacept, a CTLA4 fusion protein, was reported to be beneficial in CTLA4 haploinsufficiency patients with autoimmune cytopenias, as well as lung, gut, and central nervous system involvement (Schwab et al. 2018; Egg et al. 2021).
There are currently 45 known genes associated with immune dysregulation. For patients with common variable immunodeficiency (CVID; diagnosis based on clinical picture and immune laboratory values) or autoimmune lymphoproliferative syndrome (ALPS), pursuing a genetic diagnosis of CTLA4 haploinsufficiency has a direct impact on affected individuals due to available treatment with Abatacept.
Here, we describe a novel splice site CTLA4 mutation, resulting in a phenotype of various autoimmune manifestations and lymphocytic interstitial pneumonia with almost no history of infections.
Case Presentation
Clinical case
The proband is a 24-year-old female, born to non-consanguineous parents of East Asian descent. Family history was significant for the patient’s father who passed away from metastatic adrenal carcinoma; however, there is no other family history suggestive of immune deficiency, immune dysregulation, nor other malignancies.
She first presented with Hashimoto’s thyroiditis at 11 years, followed by Coombs positive autoimmune hemolytic anemia (AIHA) and thrombocytopenia over the next two years. She required blood transfusions and was treated with a single infusion of intravenous immunoglobulins as well as a course of Prednisone. With regards to infections, four years after her initial presentation she developed Salmonella bacteremia and osteomyelitis of the left distal femur while being on Prednisone, with no other bacterial, viral or opportunistic infections since then. At 17 years of age, she was admitted for fever and diffuse lymphadenopathy. Her work up revealed multiple pulmonary nodules on MRI, later diagnosed as lymphocytic interstitial pneumonia and follicular bronchiolitis. Other immune dysregulation features included alopecia areata, mild eczema managed topically, and photosensitive skin rash.
Over the years she received the diagnosis of ALPS-like syndrome until genetic investigation revealed her diagnosis. For the cytopenias, she continued treatment with steroids and Mycophenolate Mofetil with partial response to this date.
Immune evaluation
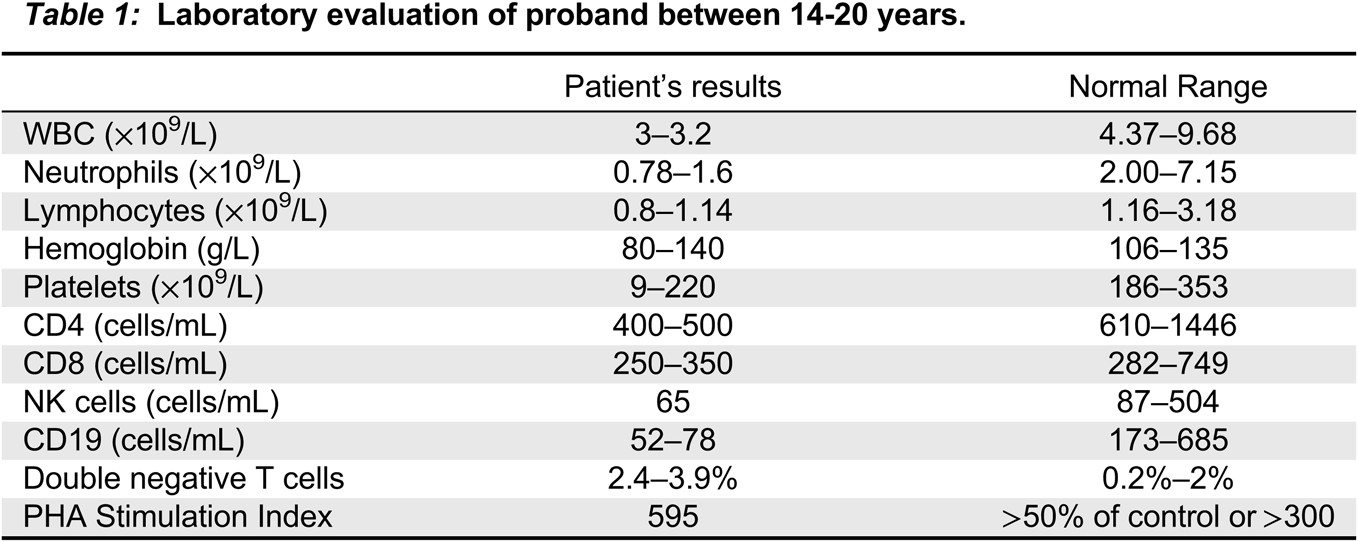
The patient’s immune work up (Table 1) was remarkable for leukopenia 3−3.2 (normal: 4.37−9.68 × 109/L), neutropenia 0.78−1.6 (normal: 2.00−7.15 × 109/L), persistent lymphopenia 0.8−1.14 (normal: 1.16−3.18 × 109/L), hemoglobin 80−140 (normal: 106−135 g/L), and platelets 9−220 (normal: 186−353 × 109/L). Immunoglobulins were within the normal range and specific vaccine titers were protective. Lymphocyte subsets showed CD4, CD8, B cell, and NK cell lymphopenia. T cell function was normal, including normal PHA stimulation index and robust response to antigen stimulation to Candida, varicella-zoster, herpes simplex and CMV. CD45 RA/RO showed increased amount of memory T cells compared to control (not shown). Autoantibodies including RF, anti-dsDNA, anti-ENA, and ANCA were negative although ANA was briefly positive (1:160) with normal C3 and C4.
Table 1:
Genetic investigations
Initial genetic analysis with an ALPS panel was returned negative. Research whole exome sequencing (WES) analysis subsequently revealed a novel heterozygous variant in CTLA4 (NM_005214.5), c.457+5delG, which was not previously found in large population databases including gnomAD nor dbSNP. The variant is an intronic change located 5 bases distal to the exon in a splice site, predicted in-silico to cause skipping of exon 2 of the CTLA4 gene and loss-of-function. The variant was subsequently confirmed with a clinical PID panel (Prevention Genetics).
Discussion
CTLA4 haploinsufficiency, first described in 2014, is caused by heterozygous variants in CTLA4 and characterized by a variety of clinical manifestations including hypogammaglobulinemia, T cell lymphopenia, autoimmune cytopenias, and lymphocytic infiltration of non-lymphoid organs as well as malignancy. Mutations described have thus far included nonsense, missense, frame-shift and splice site mutations and were shown to cause reduction in CTLA4 mRNA and protein expression (Schubert et al. 2014; Sun et al. 2014; Hayakawa et al. 2016; Mahat et al. 2021). Importantly, human biallelic CTLA4 deficiency has not yet been described, and likely indicates incompatibility with life.
The CTLA4 gene contains 4 exons encoding the signal peptide (exon 1), ligand binding and dimerization domains (exon 2), transmembrane domain (exon 3), and cytoplasmic tail (exon 4). Genetic aberrations targeting the signal peptide have been shown to abolish CTLA4 protein expression (Schubert et al. 2014), while those affecting exon 2 impair protein dimerization and interactions with the CD80 and CD86 co-stimulatory ligands (Schubert et al. 2014; Schwab et al. 2018). Mutations in exon 3 impair ligand binding and uptake through the CTLA4 transendocytosis pathway (Schubert et al. 2014; Schwab et al. 2018).
Although protein and mRNA expression of CTLA4 in Treg cells were not performed in our patient, in-silico predictions indicate that the c.457+5delG variant most probably leads to a splice anomaly and reduced CTLA4 expression. The clinical presentation of this patient was relatively similar to previously reported cases of autosomal dominant CTLA4 haploinsufficiency. Furthermore, CTLA4 was the only candidate gene identified on WES to explain this patient’s clinical manifestations. In addition, this variant was not reported in large population databases, increasing our confidence in this variant as accountable for the immune dysregulation seen in our patient.
In most of the reported cases, patients were diagnosed initially with CVID, while further genetic analysis showed variants in CTLA4 (Schubert et al. 2014; Sun et al. 2014; Mahat et al. 2021). Clinically, our patient was followed for years with a diagnosis of ALPS-like syndrome and treated empirically with immunosuppression and immune modulators according to symptoms. ALPS is a rare condition, characterized by lymphoproliferation, autoimmune manifestations, and susceptibility to malignancy. ALPS and CVID may overlap in some of the clinical and immunological features such as autoimmunity, lymphoproliferation, and susceptibility to malignancy. Often times patients receive a diagnosis of ALPS or CVID without additional genetic investigation due to low yield of genetic testing, furthermore, requesting genetic testing as part of the immunological workup was not recommended in previous years, especially in cases without a clear family history of immunodeficiency (Cunningham-Rundles 2010; Rosenzweig et al. 2016). In this case, further genetic analysis was able to elucidate the underlying diagnosis and provide specific treatment option with Abatacept.
REFERENCES
Cunningham-Rundles, C. 2010. How I treat common variable immune deficiency. Blood, 116(1): 7 [online]. Available from /pmc/articles/PMC2904582/ [accessed 5 April 2022].
Egg D., Rump I.C., Mitsuiki N., Rojas-Restrepo J., Maccari M.E., Schwab C., Gabrysch A., Warnatz K., Goldacker S., Patiño V., Wolff D., Okada S., Hayakawa S., Shikama Y., Kanda K., Imai K., Sotomatsu M., Kuwashima M., Kamiya T., Morio T., Matsumoto K., Mori T., Yoshimoto Y., Dybedal I., Kanariou M., Kucuk Z.Y., Chapdelaine H., Petruzelkova L., Lorenz H.M., Sullivan K.E., Heimall J., Moutschen M., Litzman J., Recher M., Albert M.H., Hauck F., Seneviratne S., Schmid P.J, Kolios A., Unglik G., Klemann C., Snapper S., Giulino-Roth L., Svaton M., Platt C.D., Hambleton S., Neth O., Gosse G., Reinsch S., Holzinger D., Kim Y.J., Bakhtiar S., Atschekzei F., Schmidt R., Sogkas G., Chandrakasan S., Rae W., Derfalvi B., Marquart H.V., Ozen A., Kiykim A., Karakoc-Aydiner E., Králíčková P., de Bree G., Kiritsi D., Seidel M.G., Kobbe R., Dantzer J., Alsina L., Armangue T., Lougaris V., Agyeman P., Nyström S., Buchbinder D., Arkwright P.D., and Grimbacher B. 2021. Therapeutic options for CTLA-4 insufficiency. Trans. Clinical Immunol. 149(2): 736–746. [accessed 5 April 2022].
Hayakawa S., Okada S., Tsumura M., Sakata S., Ueno Y., Imai K., Morio T., Ohara O., Chayama K., and Kobayashi M. 2016. A Patient with CTLA-4 Haploinsufficiency Presenting Gastric Cancer. J. Clinical Immunol. 36(1): 28–32.
Kuehn H.S., Ouyang W., Lo B., Deenick E.K., Niemela J.E., Avery D.T., Schickel J-N., Tran D.Q., Stoddard J., Zhang Y., Frucht D.M., Dumitriu B., Scheinberg P., Folio L.R., Frein C.A., Price S., Koh C., Heller T., Seroogy C.M., Huttenlocher A., Rao V.K., Su H.C., Kleiner D., Notarangelo L.D., Rampertaap Y., Olivier K.N., McElwee J., Hughes J., Pittaluga S., Oliveira J.B., Meffre E., Fleisher T.A., Holland S.M., Lenardo M.J., Tangye S.G., and Uzel G. 2014. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science, 345(6204): 1623–1627.
Lo, B., Zhang, K., Lu, W., Zheng, L., Zhang, Q., Kanellopoulou, C., Zhang, Y., Liu, Z., Fritz, J.M., Marsh, R., Husami, A., Kissell, D., Nortman, S., Chaturvedi, V., Haines, H., Young, L.R., Mo, J., Filipovich, A.H., Bleesing, J.J., Mustillo, P., Stephens, M., Rueda, C.M., Chougnet, C.A., Hoebe, K., McElwee, J., Hughes, J.D., Karakoc-Aydiner, E., Matthews, H.F., Price, S., Su, H.C., Rao, V.K., Lenardo, M.J., and Jordan, M.B. 2015. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (New York, N.Y.) 349(6246): 436–440 [online]. Available from https://pubmed.ncbi.nlm.nih.gov/26206937/ [accessed 30 April 2022].
Mahat, U., Ambani, N.M., Rotz, S.J., and Radhakrishnan, K. 2021. Heterozygous CTLA4 splice site mutation c.458-1G > C presenting with immunodeficiency and variable degree of immune dysregulation in three generation kindred of Caribbean descent. Pediatr. Hematol. Oncol. 38(7): 658–662 [online]. Available from https://www.tandfonline.com/action/journalInformation?journalCode=ipho20 [accessed 3 April 2022].
Rosenzweig, S., Maffucci1, P., Filion, C.A., Boisson, B., Itan, Y., Shang, L., Casanova, J.-L., and Cunningham-Rundles, C. 2016. Article 220 1 Maffucci et al. Genetic Diagnosis Using WES in CVID Frontiers in Immunology | www. 7, p. 220. [online]. Available from www.frontiersin.org.
Schubert D., Bode C., Kenefeck R., Hou T.Z., Wing J.B., Kennedy A., Bulashevska A., Petersen B.-S., Schäffer A.A., Grüning B.A., Unger S., Frede N., Baumann U., Witte T., Schmidt R.E., Dueckers G., Niehues T., Seneviratne S., Kanariou M., Speckmann C., Ehl S., Rensing-Ehl A., Warnatz K., Rakhmanov M., Thimme R., Hasselblatt P., Emmerich F., Cathomen T., Backofen R., Fisch P., Seidl M., May A., Schmitt-Graeff A., Ikemizu S., Salzer U., Franke A., Sakaguchi S., Walker L.S.K., Sansom D.M., and Grimbacher B. 2014. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20(12): 1410–1416.
Schwab C., Schwab C., Gabrysch A., Olbrich P., Patiño V., Warnatz K., Wolff D., Hoshino A., Kobayashi M., Imai K., Takagi M., Dybedal I., Haddock J.A., Sansom D.M., Lucena J.M., Seidl M., Schmitt-Graeff A., Reiser V., Emmerich F., Frede N., Bulashevska A., Salzer U., Schubert D., Hayakawa S., Okada S., Kanariou M., Kucuk Z.Y., Chapdelaine H., Petruzelkova L., Sumnik Z., Sediva A., Slatter M., Arkwright P.D., Cant A., Lorenz H-M., Giese T., Lougaris V., Plebani A., Price C., Sullivan K.E., Moutschen M., Litzman J., Freiberger T., van de Veerdonk F.L., Recher M., Albert M.H., Hauck F., Seneviratne S., Schmid J.P., Kolios A., Unglik G., Klemann C., Speckmann C., Ehl S., Leichtner A., Blumberg R., Franke A., Snapper S., Zeissig S., Cunningham-Rundles C., Giulino-Roth L., Elemento O., Dückers G., Niehues T., Fronkova E., Kanderová V., Platt C.D., Chou J., Chatila T.A., Geha R., McDermott E., Bunn S., Kurzai M., Schulz A., Alsina L., Casals F., and Deyà-Martinez A. 2018. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4–insufficient subjects. J. Allergy Clinical Immunol. 142(6): 1932–1946.
Sun H., Ouyang W., Lo B., Deenick E.K., Niemela J.E., Avery D.T., Schickel J-N., Tran D.Q., Stoddard J., Zhang Y., Frucht D.M., Dumitriu B., Scheinberg P., Folio L.R., Frein C.A., Price S., Christopher Koh S., Heller T., Seroogy C.M., Huttenlocher A., Rao V.K., Su H.C., Kleiner D., Notarangelo L.D., Rampertaap Y., Olivier K.N., McElwee J., Hughes J., Pittaluga S., Oliveira J.B., Meffre E., Fleisher T.A., Holland S.M., Lenardo M.J., Tangye S.G., and Uzel G. 2014. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4 HHS Public Access. Science, 345(6204): 1623–1627.
Tivol, E.A., Borriello, F., Schweitzer, A.N., Lynch, W.P., Bluestone, J.A., and Sharpe, A.H. 1995. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immun. 3(5): 541–547. [online]. Available from https://pubmed.ncbi.nlm.nih.gov/7584144/ [accessed 30 April 2022].
Waterhouse, P., Penninger, J.M., Timms, E., Wakeham, A., Shahinian, A., Lee, K.P., Thompson, C.B., Griesser, H., and Mak, T.W. 1995. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Science, 270(5238): 985–988 [online]. Available from https://www.science.org/doi/abs/10.1126/science.270.5238.985 [accessed 30 April 2022].
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 2 • June 2022
Pages: 40 - 44
History
Received: 3 May 2022
Accepted: 15 May 2022
Accepted manuscript online: 19 May 2022
Version of record online: 19 May 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JennyGarkaby, Laura EdithAbrego Fuentes, JessicaWillett Pachul, DanieleMerico, and LindaVong. 2022. CTLA4 haploinsufficiency caused by a novel heterozygous splice site mutation. LymphoSign Journal.
9(2): 40-44. https://doi.org/10.14785/lymphosign-2022-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
