An unusual presentation of DiGeorge syndrome
Abstract
Introduction: DiGeorge syndrome is a heterogenous disorder with various clinical presentations. Common features include thymic hypoplasia, T cell lymphopenia, conotruncal heart defects, facial dysmorphism, cleft palate, developmental delay, and hypoparathyroidism. The severity of this condition varies, however typical presentation includes congenital heart defects and characteristic facial features. Isolated hypocalcemia in DiGeorge syndrome is rarely seen in neonates but rather as the sole manifestation in older teenagers or adults.
Aim: To report a case of an atypical presentation of DiGeorge syndrome.
Results: We report here a case of an infant who was diagnosed with DiGeorge syndrome, with seizures being the only clinical manifestation displayed by the patient. He was found to have low T cell receptor excision circle levels on a newborn screen (NBS) for severe combined immunodeficiency (SCID). He did not have facial dysmorphism nor cardiac defect.
Conclusion: Our case shows that severe hypocalcemia can be the only presenting symptom in DiGeorge syndrome. Based on this case, we recommend physicians test for calcium levels and PTH at the first encounter with a patient who screened positive during NBS for SCID.
Statement of Novelty: We describe an infant with DiGeorge syndrome who presented with severe hypocalcemia.
Introduction
DiGeorge syndrome (DGS, OMIM #188400) is one of a group of phenotypically similar disorders caused by microdeletions of chromosome 22q11.2 and rarely by microdeletions in 10p, 18q21.33, or 4q21.3-q25 (Morrow et al. 2018; Greenberg et al. 1988; Fukushima et al. 1992). Genes mapping to the deleted 22q11.2 region include TBX1 (Chieffo et al. 1997), COMT (Shashi et al. 2006), ZNF74 (Aubry et al. 1993), and DGCR6 (Demczuk et al. 1996). The related syndromes include velocardiofacial syndrome (Shprintzen syndrome) (Shprintzen et al. 1978), and conotruncal anomaly face (CTAF) syndrome (Burn et al. 1993). About 10% of cases are transmitted in an autosomal dominant fashion but most are generated de novo (McDonald-McGinn et al. 2001). The group of disorders are currently referred to by some as 22q11.2 deletion syndrome but the more traditional term DGS is still used, especially when the immune system is abnormal (Burn 1999).
The syndrome is heterogenous and includes most frequently thymic hypoplasia, congenital heart defects (ventricular septal defect, Truncus arteriosus, Tetralogy of Fallot), facial anomalies (including cleft palate, epicanthus, telecanthus, wide and prominent nasal bridge, bulbous nose and low set ears), and developmental delay (Botto et al. 2003; Goodship et al. 1998). Hypoparathyroidism, hearing impairment, and absent dentition are less common but still a frequent feature of the syndrome. The 22q11.2 microdeletion causes a fault in the development of cervical neural migration in the derivatives of the pharyngeal arches, thus explaining the poor development of the thymus and parathyroid glands (Molsted et al. 2010).
The extent of immunodeficiency, dysmorphic features, and hypoparathyroidism is highly variable. Yet in most instances patients have a combination of features which alerts healthcare providers to the possible diagnosis of DGS.
We report here a rare case of an infant who was found to have low TRECs in a newborn screen (NBS) for severe combined immunodeficiency (SCID). He had no facial dysmorphism nor cardiac lesion. He was admitted for seizures at 3 weeks of age.
Case presentation
The patient, a product of non-consanguineous parents, was delivered at 37 weeks gestation via elective c-section due to maternal gestational diabetes and hypertension. The mother was treated with Labetolol and low dose aspirin since week 34 of gestation. The delivery was uncomplicated but the patient experienced hypoglycemia after birth which resolved within days, as well as a rise in bilirubin and jaundice which resolved in a week.
The patient has 2 healthy siblings. There is no history of immunodeficiency or severe infections in family members. NBS for SCID revealed low T cell receptor excision circle (TREC) levels in the patient’s blood sample (53, cut-off: 75). This result triggered a retrieval visit with a NBS genetic counselor and Immunology staff (Reid et al. 2017).
On examination, the baby appeared alert with proper muscle tone and was positive for moro, stepping, and grasp reflex. He had no notable facial abnormalities with the exception of mild ear lobe folding. No heart murmurs were noted on auscultation. The rest of the physical assessment was normal.
The parents reported he was feeding well with a combination of breastmilk and formula. According to NBS protocol, we sent out the following tests: repeat TREC, CBC and differential, immunophenotyping, 22q11.2 FISH, lymphocyte proliferation to phytohemagglutinin (PHA), and CMV status in baby and mom. A genetic panel for primary immunodeficiency as well as samples for research (DNA for future whole exome sequencing if needed) were also obtained. Lymphocyte count was within normal limits but immunophenotyping revealed low CD3+, CD4+, and CD8+ cells with increased CD19+ B cells and CD16+CD56+ NK cells. However, T cell function appeared normal with a PHA response within normal range (Table 1).
Table 1:
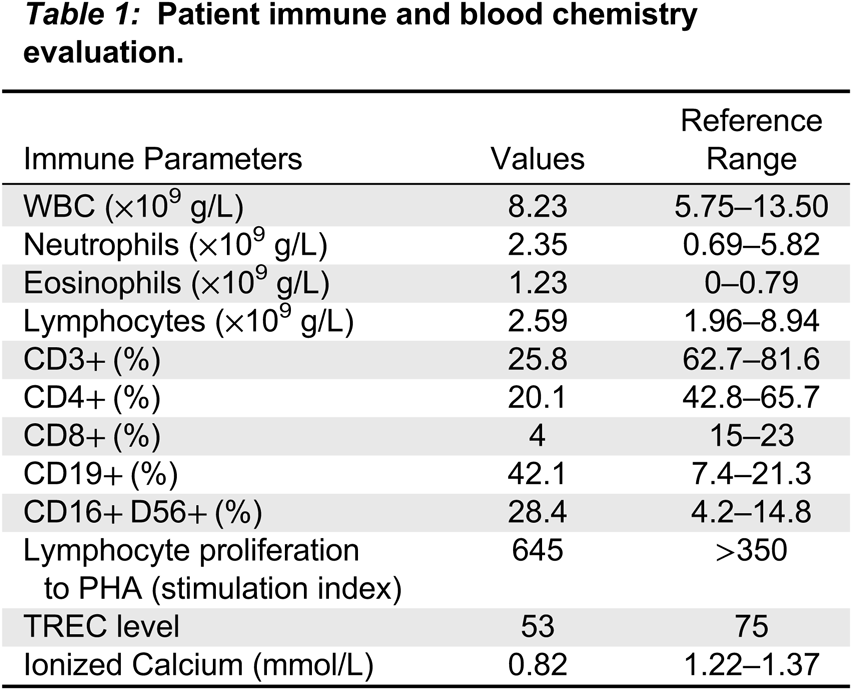
He continued to feed well and gain weight until he was 5 weeks old when he was admitted to hospital with status epilepticus secondary to profound hypocalcemia. One day before admission his FISH analysis was reported confirming a deletion in 22q11.2. He gradually improved in hospital with calcium supplementation and anti-seizure medications including Keppra. He had no seizure activity and a normal EEG after cessation of Keppra. However, this was re-started following MRI findings of multiple anomalies consistent with DGS including diffuse polymicrogyria, heterotopia, and abnormal semicircle canals, as well as a left frontal subdural hematoma.
Discussion
Newborn screening for SCID revolutionized the field of primary immunodeficiency (Kwan et al. 2014). Using TRECs as a surrogate indicator of extremely low circulating lymphocytes, mainly naïve (new thymic emigrants), has allowed for effective detection of typical SCID (Puck 2012). Still, atypical SCID such as Zap-70 deficiency or leaky SCIDs may be missed by this tool (Grazioli et al. 2014; Kwan et al. 2014; Kwan and Puck 2015). In contrast, other non-SCID immunodeficiencies have been picked up as incidental findings by this method. While the list of these conditions continues to grow, most commonly detected are cases with DiGeorge syndrome, Ataxia Telangiectasia, and FOXN1-related lymphopenia (Scott et al. 2021; Mandola et al. 2019; Barry et al. 2017). However, the majority of NBS-positive cases appear insignificant (prematurity) or secondary to maternal or perinatal events (such as maternal or newborn treatment with immunosuppressive medications or perinatal stress events) (Mandola et al. 2019).
The TREC levels in typical SCID are traditionally low or undetectable. That was not the case in the patient reported here, where levels were only somewhat lower than the cut-off level. This and the lower than normal level of CD3+ cells could have been associated with a putative T cell immunodeficiency or secondary to the stress experienced by this baby. Moreover, the normal response to the mitogen PHA eliminated most cases of profound T cell immunodeficiency.
As part of our routine first visit evaluation (Biggs et al. 2017; Reid et al. 2017), we requested FISH analysis with the intent to rule out DGS, one of the causes of neonatal lymphopenia. While in retrospective analysis, an acceptable option for diagnosis, the finding of microdeletion of 22q11.2 was surprising given this case had no obvious dysmorphic facial features nor cardiac defects.
Our patient did not display any clinical symptoms until age 3 weeks when seizures began. Hypocalcemia was confirmed in hospital, 20 hr after the genetic FISH analysis was released. Indeed, sudden seizures were the only clinical manifestation displayed by the patient.
Frequently in newborns and infants, hypocalcemia may be present in combination with other features associated with DGS (Barry et al. 2017). Rarely, hypocalcemia presents as the sole manifestation in older teenagers of adults (Cabrer et al. 2018; Zammit et al. 2013; Maalouf et al. 2004). Few case reports in adults identified retrospectively facial anomalies typical of DGS (Vogels et al. 2014). Our case demonstrates that severe hypocalcemia can be the only clinical manifestation of DGS and subsequently we recommend testing for calcium levels and PTH at the first encounter with a positive NBS for SCID.
REFERENCES
Aubry M., Demczuk S., Desmaze C., Aikem M., Aurias A., Julien J.P., and Rouleau G.A. 1993. Isolation of a zinc finger gene consistently deleted in DiGeorge syndrome. Hum. Mol. Genet. 2: 1583–7.
Barry J.C., Crowley T.B., Jyonouchi S., Heimall J., Zackai E.H., Sullivan K.E., and Mcdonald-Mcginn D.M. 2017. Identification of 22q11.2 Deletion Syndrome via Newborn Screening for Severe Combined Immunodeficiency. J. Clin. Immunol. 37: 476–85.
Biggs C.M., Haddad E., Issekutz T.B., Roifman C.M., and Turvey S.E. 2017. Newborn screening for severe combined immunodeficiency: a primer for clinicians. CMAJ, 189: E1551–7.
Botto L.D., May K., Fernhoff P.M., Correa A., Coleman K., Rasmussen S.A., Merritt R.K., O’leary L.A., Wong L.Y., Elixson E.M., Mahle W.T., and Campbell R.M. 2003. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics, 112: 101–7.
Burn J. 1999. Closing time for CATCH22. J. Med. Genet. 36: 737–8.
Burn J., Takao A., Wilson D., Cross I., Momma K., Wadey R., Scambler P., and Goodship J. 1993. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J. Med. Genet. 30: 822–4.
Cabrer M., Serra G., Gogorza M.S., and Pereg V. 2018. Hypocalcemia due to 22q11.2 deletion syndrome diagnosed in adulthood. Endocrinol. Diabetes Metab. Case Rep. 2018: 17-0140.
Chieffo C., Garvey N., Gong W., Roe B., Zhang G., Silver L., Emanuel B.S., and Budarf M.L. 1997. Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics, 43: 267–77.
Demczuk S., Thomas G., and Aurias A. 1996. Isolation of a novel gene from the DiGeorge syndrome critical region with homology to Drosophila gdl and to human LAMC1 genes. Hum. Mol. Genet. 5: 633–8.
Fukushima Y., Ohashi H., Wakui K., Nishida T., Nakamura Y., Hoshino K., Ogawa K., and Oh-Ishi T. 1992. DiGeorge syndrome with del(4)(q21.3q25): possibility of the fourth chromosome region responsible for DiGeorge syndrome. (Abstract). Am. J. Hum. Genet, 51 (suppl.): A80.
Goodship J., Cross I., Liling J., and Wren C. 1998. A population study of chromosome 22q11 deletions in infancy. Arch. Dis. Child. 79: 348–51.
Grazioli S., Bennett M., Hildebrand K.J., Vallance H., Turvey S.E., and Junker A.K. 2014. Limitation of TREC-based newborn screening for ZAP70 Severe Combined Immunodeficiency. Clin. Immunol. 153: 209–10.
Greenberg F., Elder F.F., Haffner P., Northrup H., and Ledbetter D.H. 1988. Cytogenetic findings in a prospective series of patients with DiGeorge anomaly. Am. J. Hum. Genet. 43: 605–11.
Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., Baker M., Ballow M., Bartoshesky L.E., Bonilla F.A., Brokopp C., Brooks E., Caggana M., Celestin J., Church J.A., Comeau A.M., Connelly J.A., Cowan M.J., Cunningham-Rundles C., Dasu T., Dave N., De La Morena M.T., Duffner U., Fong C.T., Forbes L., Freedenberg D., Gelfand E.W., Hale J.E., Hanson I.C., Hay B.N., Hu D., Infante A., Johnson D., Kapoor N., Kay D.M., Kohn D.B., Lee R., Lehman H., Lin Z., Lorey F., Abdel-Mageed A., Manning A., Mcghee S., Moore T.B., Naides S.J., Notarangelo L.D., Orange J.S., Pai S.Y., Porteus M., Rodriguez R., Romberg N., Routes J., Ruehle M., Rubenstein A., Saavedra-Matiz C.A., Scott G., Scott P.M., Secord E., Seroogy C., Shearer W.T., Siegel S., Silvers S.K., Stiehm E.R., Sugerman R.W., Sullivan J.L., Tanksley S., Tierce M.L.T., Verbsky J., Vogel B., Walker R., Walkovich K., Walter J.E., Wasserman R.L., Watson M.S., Weinberg G.A., Weiner L.B., Wood H., Yates A.B., Puck J.M., and Bonagura V.R. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA, 312: 729–38.
Kwan A. and Puck J.M. 2015. History and current status of newborn screening for severe combined immunodeficiency. Semin Perinatol, 39: 194–205.
Maalouf N.M., Sakhaee K., and Odvina C.V. 2004. A case of chromosome 22q11 deletion syndrome diagnosed in a 32-year-old man with hypoparathyroidism. J. Clin. Endocrinol. Metab. 89: 4817–20.
Mandola A.B., Reid B., Sirror R., Brager R., Dent P., Chakroborty P., Bulman D.E., and Roifman C.M. 2019. Ataxia Telangiectasia Diagnosed on Newborn Screening-Case Cohort of 5 Years’ Experience. Front. Immunol. 10: 2940.
Mcdonald-Mcginn D.M., Tonnesen M.K., Laufer-Cahana A., Finucane B., Driscoll D.A., Emanuel B.S., and Zackai E.H. 2001. Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet Med. 3: 23–9.
Molsted K., Boers M., and Kjaer I. 2010. The morphology of the sella turcica in velocardiofacial syndrome suggests involvement of a neural crest developmental field. Am. J. Med. Genet. 152A: 1450–7.
Morrow B.E., Mcdonald-Mcginn D.M., Emanuel B.S., Vermeesch J.R., and Scambler P.J. 2018. Molecular genetics of 22q11.2 deletion syndrome. Am. J. Med. Genet. 176: 2070–81.
Puck J.M. 2012. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J. Allergy Clin. Immunol. 129: 607–16.
Reid B.E., Ovadia A., and Dinur Schejter Y. 2017. Managing Newborn Screening for SCID in a Referral Centre. LymphoSign J. 4(2).
Scott O., Garkaby J., Willett-Pachul J., Mandola A.B., and Pasternak Y. 2021. A novel splice site variant in FOXN1 in a patient with abnormal newborn screening for severe combined immunodeficiency and congenital lymphopenia. LymphoSign Journal, 8: 1–4.
Shashi V., Keshavan M.S., Howard T.D., Berry M.N., Basehore M.J., Lewandowski E., and Kwapil T.R. 2006. Cognitive correlates of a functional COMT polymorphism in children with 22q11.2 deletion syndrome. Clin. Genet. 69: 234–8.
Shprintzen R.J., Goldberg R.B., Lewin M.L., Sidoti E.J., Berkman M.D., Argamaso R.V., and Young D. 1978. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft. Palate. J. 15: 56–62.
Vogels A., Schevenels S., Cayenberghs R., Weyts E., Van Buggenhout G., Swillen A., Van Esch H., De Ravel T., Corveleyn P., and Devriendt K. 2014. Presenting symptoms in adults with the 22q11 deletion syndrome. Eur. J. Med. Genet. 57: 157–62.
Zammit A., Grech Marguerat D., Psaila J., and Attard A. 2013. DiGeorge Syndrome Presenting as Hypocalcaemia-Induced Seizures in Adulthood. Clin. Med. Case Rep. 2013: 1–4.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 2 • June 2022
Pages: 52 - 56
History
Received: 9 May 2022
Accepted: 22 May 2022
Accepted manuscript online: 24 May 2022
Version of record online: 24 May 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JennyGarkaby, Laura EdithAbrego Fuentes, JessicaWillett Pachul, AbbyWatts-Dickens, and MeghanFraser. 2022. An unusual presentation of DiGeorge syndrome. LymphoSign Journal.
9(2): 52-56. https://doi.org/10.14785/lymphosign-2022-0005
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Management of newborn screening for severe combined immunodeficiency at a quaternary referral centre—an updated algorithm
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
