Primary antibody deficiency associated with ring chromosome 18
Abstract
Background: Patients with chromosome 18 abnormalities can present with an immune phenotype that resembles common variable immunodeficiency. Knowledge of the genes underlying the immune defects related to chromosome 18 aberrations could improve our understanding of the molecular basis of primary antibody deficiencies. Here we present a patient with ring chromosome 18 affected by primary antibody deficiency and autoimmunity.
Methods: Lymphocyte populations were determined by flow cytometry. Specific antibody response to protein vaccines and pneumococcal capsule antigen were measured by ELISA. Genome sequencing was performed using a PCR-free protocol.
Case: The patient was diagnosed with ring chromosome 18 for delayed growth and dysmorphic features at the age of 1 month. Array comparative genomic hybridization showed deletions of 18p11.21-pter and 18q21.31-qter. At the age of 10 months, she started having recurrent episodes of otitis media and pneumonia, as well as autoimmune arthritis. Serum immunoglobulins and specific antibody levels were low. The CD19+CD27+ memory B cell and CD45RO+ T cell populations were decreased. Recurrent infections were controlled with parenteral immunoglobulin and autoimmune arthritis was treated with systemic and intra-articular therapies.
Conclusions: Selective IgA deficiency is the most common form of immunodeficiency associated with chromosome 18 abnormalities, however patients with ring chromosome 18 may also be affected by specific antibody deficiency and require immunoglobulin replacement for optimal care. These patients might partially share the same genomic loss as in patients with non-syndromic primary antibody deficiency.
Statement of novelty: This report highlights an important teaching point about immune deficiency in a chromosomal anomaly that is not infrequently encountered in pediatric hospitals. Furthermore, our investigations provide more insight into the pathogenesis of immunodeficiency among patients with chromosome 18 abnormalities.
Introduction
Antibody deficiencies comprise the most common form of inborn errors of the immune system. Although some monogenic primary antibody deficiencies have been identified, the genetic basis is not yet clear for the majority of those patients (Conley 2009). This reflects the heterogeneous nature of immune system development and maturation. In addition to single gene defects, chromosomal structural abnormalities have been associated with primary antibody deficiencies (Cunningham-Rundles 2012; Schatorje et al. 2016). These include both autosomal and sex chromosome aneuploidies, duplication, deletions and ring chromosomes. Notably, defective humoral immunity has been described among the clinical findings of patients with different chromosome 18 abnormalities (Feingold and Schwartz 1968; Fischer et al. 1970; Michaels et al. 1971; Faed et al. 1972; Schinzel et al. 1974). Patients with deletions on both 18p and 18q have presented with selective IgA deficiency (SIgAD), hypogammaglobulinemia, impaired specific antibody responses, and loss of memory B cells (Browning 2010; Cody et al. 2014; Hasi-Zogaj et al. 2015; Calvo Campoverde et al. 2016). Additionally, deletions of chromosome 18 are often marked with severe forms of autoimmunity (Dacou-Voutetakis et al. 1999; Hasi-Zogaj et al. 2015). The combination of antibody deficiency and autoimmunity is a common feature of many patients with common variable immunodeficiency (CVID). Therefore, exploring the immunogenetics of patients with chromosome 18 abnormalities might help finding new insights into the yet-obscure genetics of primary antibody deficiencies in non-syndromic patients.
Ring chromosome 18 is characterized by dysmorphic features such as microcephaly, hypertelorism, epicanthal folds, micrognathia, and short tapering fingers. Additionally, the patients are affected by severe intellectual delay, hypotonia, seizures, white matter abnormalities, hearing loss, and growth hormone deficiency (Carter et al. 2015). Here, we report a case of ring chromosome 18 who presented with recurrent bacterial infections, low serum IgG and IgM levels, defective specific antibody responses, and severe autoimmunity. To further investigate this patient, we detected the breakpoints by array comparative genomic hybridization (aCGH) and performed genome sequencing (GS) to examine possible gene defects that could lead to the immune phenotype.
Materials and methods
Patient
The study was approved by the Research Ethics Boards (REBs) of the McGill University Health Center under the Canadian Primary Immunodeficiency Evaluation Study (CPRIMES). Written consent was obtained for the genetic and immunological investigations from the patient’s parents.
Flow cytometry
Lymphocyte immunophenotyping was perform by standard flow cytometry using antibodies, all from BD Bioscience: CD45RA/CD45RO/CD3/CD4 (cat #340571) and CD45RA/CD45RO/CD3/CD4 (cat #340574), PerCp-Cy5.5 conjugated CD19 (cat #340951), FITC conjugated IgD (cat #555778), PE conjugated IgM (cat #55579), APC conjugated CD27 (cat #337169).
Specific antibody response
Serum levels of anti-pneumococcal capsular antigen IgG for 14 pneumococcal serotypes (1, 3, 4, 6B, 7F, 9V, 11A, 12F, 14, 15B, 18C, 19F, 23F, and 33F), were determined by ELISA as previously described (Lejtenyi and Mazer 2008). Intravenous immunoglobulin (IVIG) treatment was held for 6 months prior to immunization of the patient with a 23-multivalent pneumococcal polysaccharide vaccine. Post pneumococcal antibodies levels were measured 4 weeks after immunization.
Array comparative genomic hybridization (aCGH)
Genomic copy number variants were detected in a local diagnostic laboratory, by NimbleGen CGX-12 microarray, containing 135K oligos (Signature Genomic Laboratories, Spokane, WA, USA).
DNA extraction, whole genome sequencing and data analysis
DNA was isolated from peripheral blood monocytes using a commercially available kit (Qiagen, Toronto, ON, Canada) according the manufacturer’s instructions. GS was performed on 1 μg of genomic DNA using a PCR-free protocol on an Illumina HiSeq X10 platform with 151-bp paired-end reads and a sequencing depth of >30X at the McGill University and Genome Quebec Innovation Center. Data analysis followed the Genome Analysis toolkit (GATK) best practices guidelines (https://software.broadinstitute.org/gatk/best-practices/). Copy number variants were detected using PopSV (Monlong et al. 2018).
Case summary
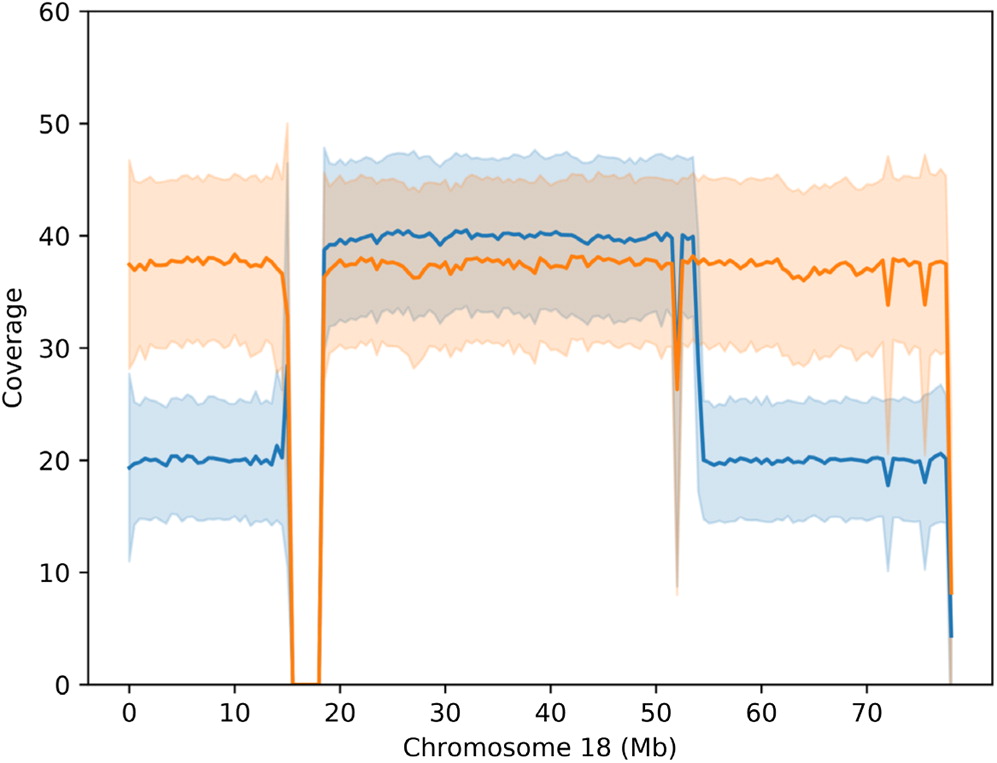
Our patient—who is currently 20 years old—was born at 36 weeks of gestation by breech vaginal delivery to non-consanguineous French-Canadian parents, after an uncomplicated pregnancy. The Apgar score was 6 and 7 at 1 and 5 minutes of life, respectively. Growth parameters at birth were weight 2530 g, length 48 cm and head circumference 31.5 cm, all over 50th percentile for gestational age. She was treated with oxygen and antibiotics for meconium aspiration. She is the youngest of 3 and her older siblings are healthy. She was later re-admitted at the age of 1 month for failure to thrive and was found to have growth hormone deficiency. At that time, she had developed dysmorphic features including borderline low set ears, slightly protruding eyes, narrow palpebral fissures, prominent maxilla with Cupid’s bow upper lip and mild micrognathia. She also had a protuberant abdomen, an umbilical hernia, and a sacral dimple. A brain MRI revealed delayed cerebral myelination that has been previously reported (Benini et al. 2012). A karyotype revealed ring chromosome 18. All metaphases exhibited 1 ring chromosome 18, and a small percentage of the cells had 2 ring 18 chromosomes. Results of aCGH showed a de novo interstitial deletion of 13q12.12 (23 544 669–24 109 193; UCSC 2009, GRCh37/hg19 Assembly), and deletions of 18p11.32p11.21 (141 491–14 117 537) and 18q21.31q23 (54 241 048–78 013 710; UCSC 2009, GRCh37/hg19 Assembly) (Figure 1).
Figure 1:
At age of 10 months, she developed recurrent otitis media requiring bilateral pressure equalizing tube placement. The patient developed multiple bacterial infections including 2 S. pneumoniae pneumonia episodes with sepsis at age 15 and 21 months, cellulitis at age 16 months and a surgical granuloma with S. aureus superinfection at 17 months of age.
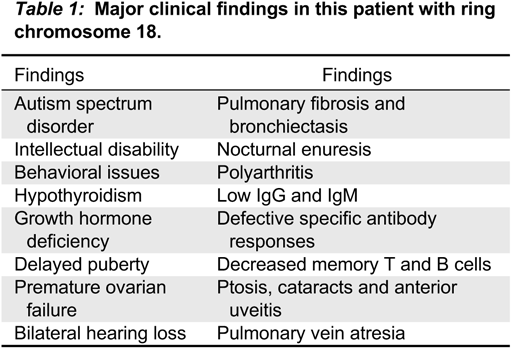
She developed hypothyroidism, anterior uveitis, and polyarthritis by the age of 2 years. She was seronegative for autoimmunity and the complement levels were normal. Her polyarthritis was controlled by systemic and intra-articular corticosteroids, Methotrexate and ultimately, Etanercept. Furthermore, she had frequent episodes of hemoptysis, secondary to complete atresia of the right upper pulmonary vein and a nearly atretic right lower pulmonary vein, which were surgically repaired. Her clinical features are summarized in Table 1.
Table 1:
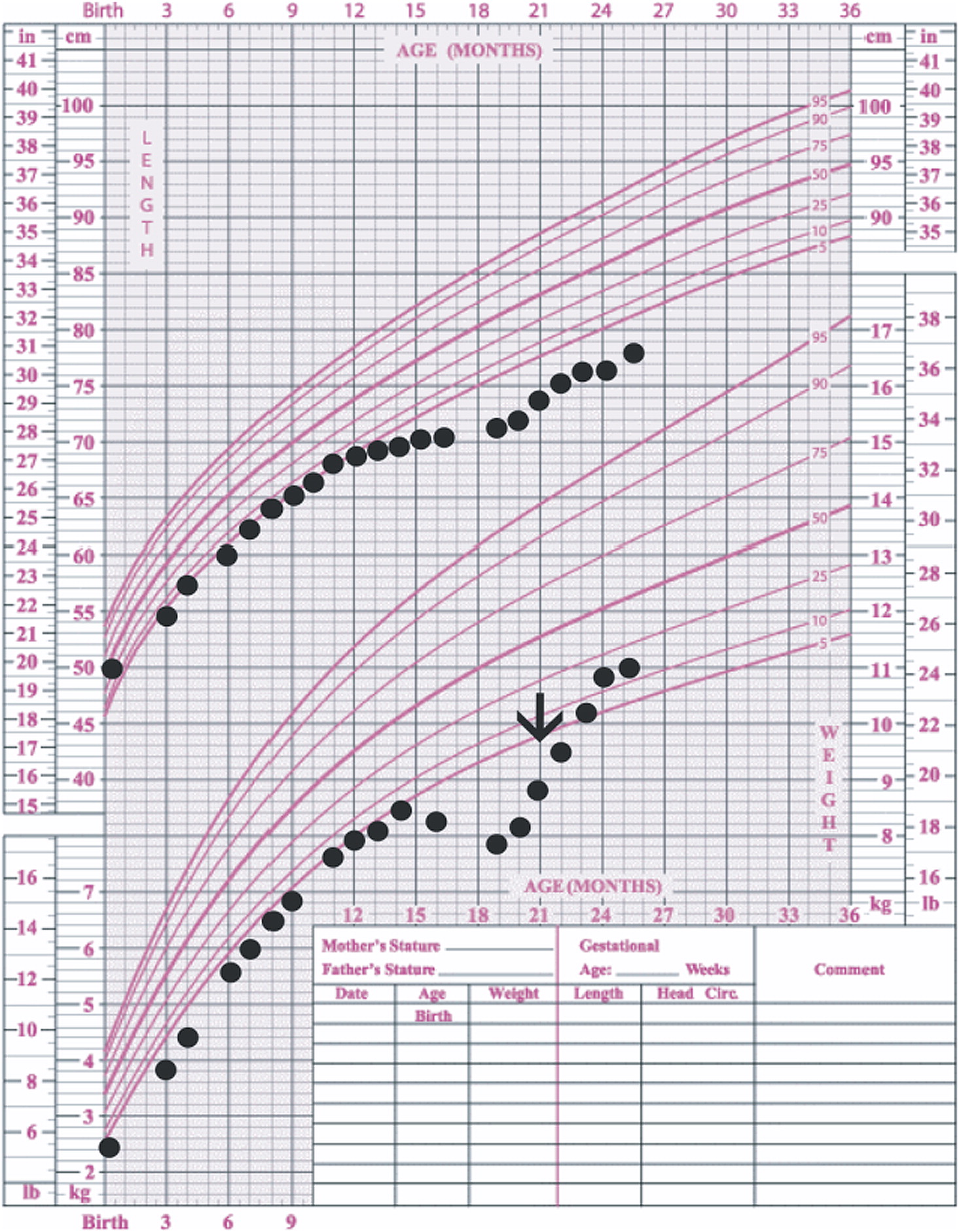
Given the recurrent infections, her immune function was assessed at age 15 months. IgG and IgM serum levels were below the normal limits for age (1.1–3.92 μg/mL and 0.12–0.85 μg/mL, respectively). She was started on monthly infusions of IVIG (600 mg/kg) at the age of 21 months and there were no further episodes of sepsis (Figure 2). Furthermore, concurrent with starting IVIG although, her height remained below the 5th percentile curve, after starting IVIG, she showed steady weight gain (Figure 3). She also received exogenous growth hormone (GH) as it had previously been shown to be beneficial in children with ring chromosome 18 and growth delay (Cody et al. 2015). A second assessment at the age of 11 years showed normal endogenous GH activity and GH was discontinued.
Figure 2:
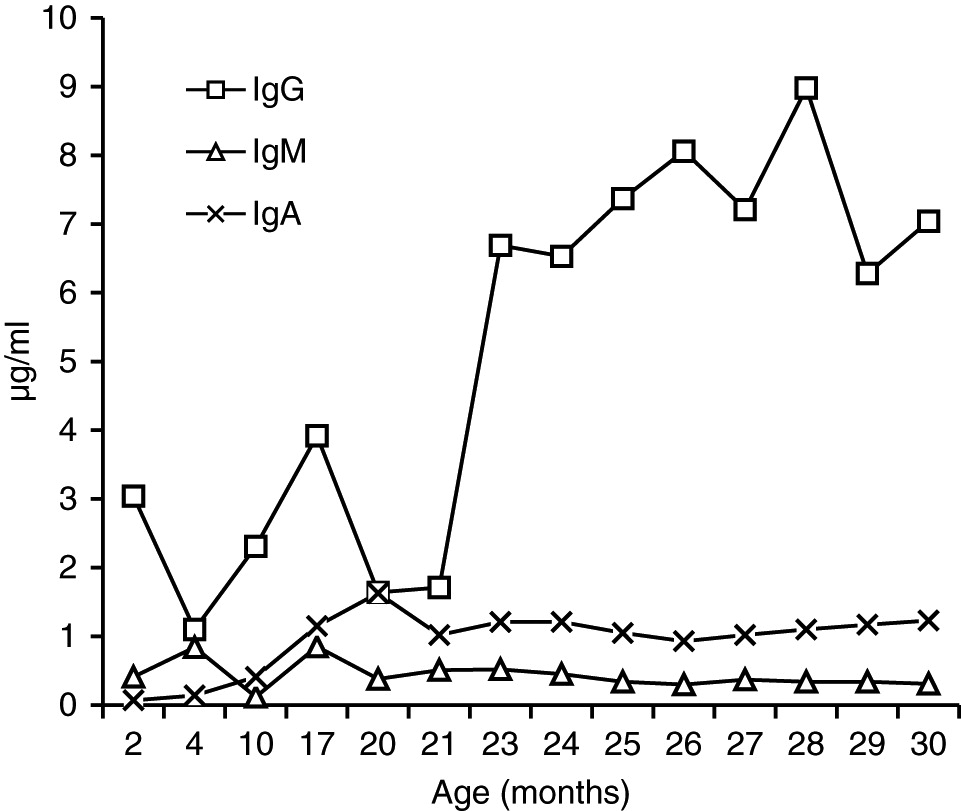
Figure 3:
In the scenario of recurrent episodes of pneumococcal infection, antibody titers to diphtheria and tetanus were obtained and were initially borderline-lower-end normal at the age of 4 months (0.3 and 0.1 IU/mL, respectively), but 6 months after a booster vaccine the serum levels were protective (2.0 IU/mL). At the age of 9 years, antibody concentrations dropped to suboptimal levels for diphtheria, tetanus, H. influenza, Mumps, Varicella zoster virus (VZV), and Rubella. Nevertheless, booster doses for diphtheria and tetanus induced again appreciable concentrations of specific antibodies (Table 2). To determine if she should remain on IVIG, we assessed specific antibody response to the 23-multivalent pneumococcal polysaccharide vaccine. Unfortunately, due to her age, pre and post pneumococcal antibodies levels were not available prior to initiation of therapy, and stored sera were not available. At the age of 9 years, IVIG was held for 6 months prior to vaccination and we measured her response 4 weeks following 23-multivalent pneumococcal polysaccharide vaccine. Her poor response confirmed the clinical suspicion of SIgAD (Table 3). During the time off IVIG therapy, she developed episodes of sinusitis, streptococcal vulvitis, and fungal onycholysis. She was restarted on immunoglobulin therapy (subcutaneous) and was also given prophylactic azithromycin.
Table 2:
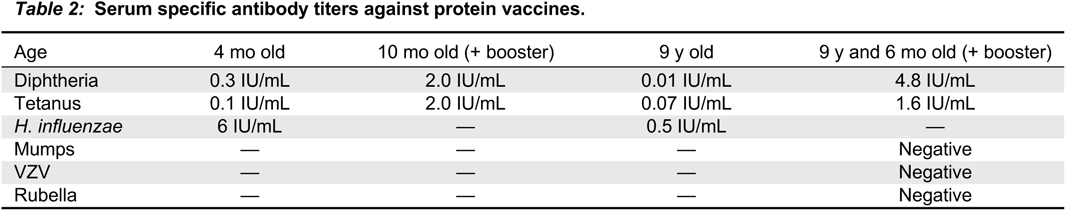
Table 3:
Note: IVIG treatment was held for 6 mo before the patient was vaccinated with a 23-multivalent pneumococcal polysaccharide vaccine. Serum antibody titers for 14 serotypes were assessed 4 wk post-immunization.
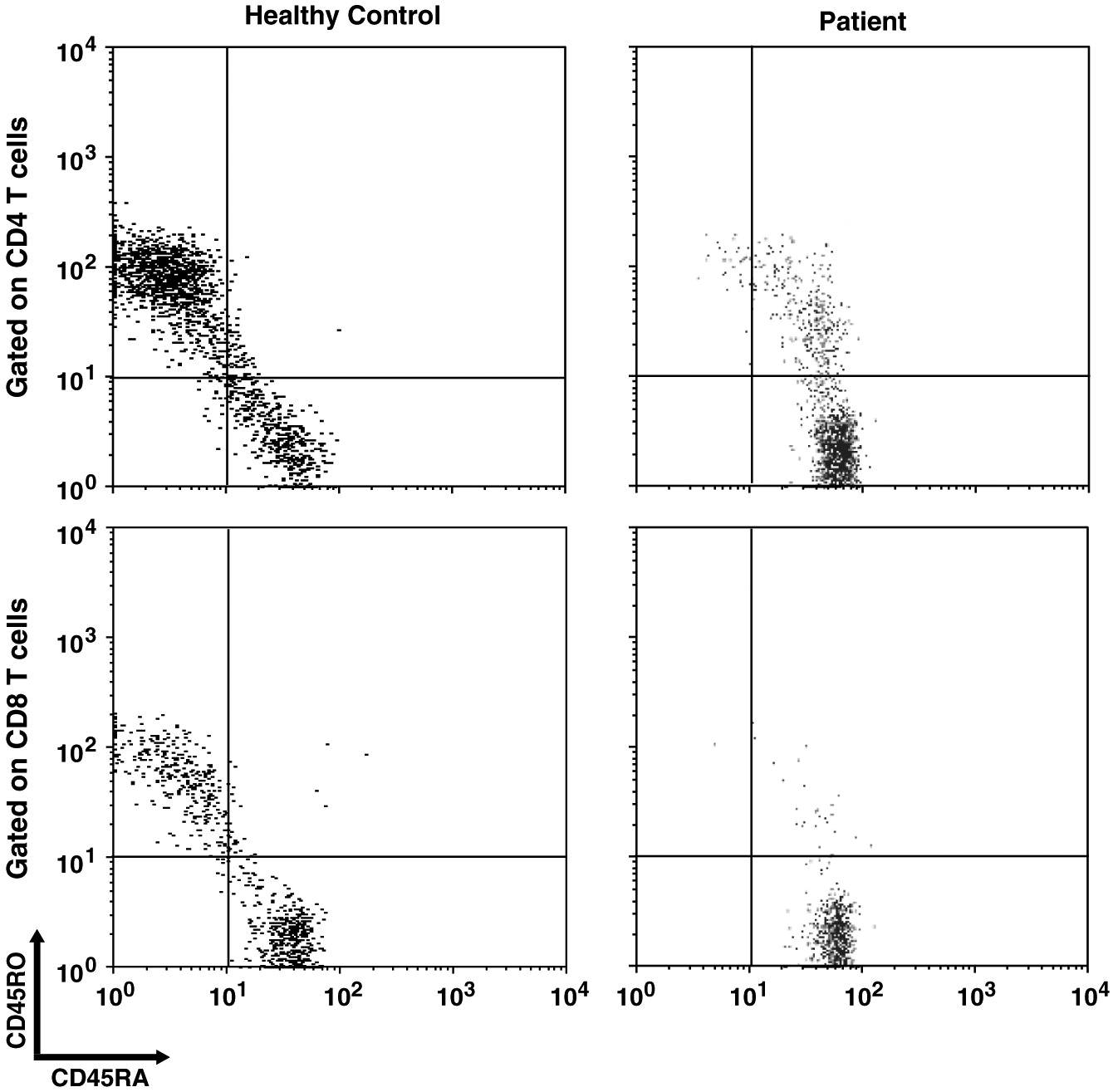
At age 13 and 18 years of age, immunophenotyping of T and B-lymphocytes revealed normal total CD4 and CD8 T cells and CD19+ B cells. However, the CD19+CD27+ memory B cell and CD19+CD27+IgD− switched memory B cell populations were decreased, compared to a health age-matched controls (Table 4). Additionally, analysis of CD45RA and RO subtypes revealed that both CD4+CD45RO+ and CD8+CD45RO+ populations were significantly decreased (Table 4) with few single positive CD45RO+ cells and approximately 5% of T cells co-expressed CD45RA and RO (Figure 4). Nevertheless, T cell function tests including phytohemagglutinin-induced proliferation and V-beta repertoire were normal (data not shown).
Table 4:
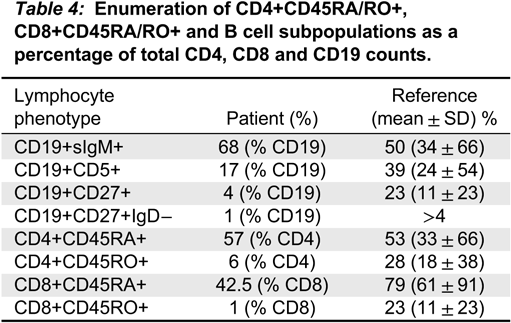
Figure 4:
To investigate possible gene variants on the non-affected sister chromosome, we performed GS. This approach was chosen to find putative variants affecting coding or intronic sequences, splice sites, and regulatory regions. The results confirmed the breakpoints at chr18:5001–15 190 000 and chr18:54 215 001–78 020 000 (UCSC 2009, GRCh37/hg19 Assembly). No pathogenic or likely pathogenic variant of the genes in the deleted regions of chromosome 18 was filtered out. Meanwhile, we questioned whether a smaller intragenic copy number variant (CNV) on the regions corresponding to deletions had been missed on aCGH. Indeed, we did not observe any other CNV in the 2 regions of interest using PopSV. We, however, used the intermediate results of PopSV and looked at bins with z score ≤−3. Using this method, we found a deletion of 4 kb (chr18:60,575,00160,579,000) deep in the middle of intron 8 of PHLPP1 with almost 2.5–3 kb distance from 5′ and 3′ splicing sites. GS further excluded known monogenic defects associated with primary antibody deficiencies.
Discussion
The phenotype of individuals with a given ring chromosome can simply resemble those with terminal deletions, but without ring formation. Nevertheless, given the secondary genomic instability, clinical presentations can be highly variable (Guilherme et al. 2011). Chromosome 18 was among the first chromosomes found in humans to be affected by ring formation (Carter et al. 2015). Although deletions of 2 regions on 18q (17 000 000–19 667 062 and 45 578 734–46 739 965) are reportedly fatal, large deletions of chromosome 18 are thought to be less lethal due to the low gene density per Mb (Cody et al. 2015). Deletions of 18q are thought to have a more variable and unpredictable phenotype compared to 18p deletions. This is likely due to the high variability of hemizygosities as well as other factors such as duplications, somatic mosaicism and ring instability (Cody et al. 2009; Carter et al. 2015; Hasi-Zogaj et al. 2015). Our patient also has a 500 kb hemizygous deletion on chromosome 13 that does not encompass any UCSC genes.
13% of patients with 18p- have low serum immunoglobulin levels. SIgAD is the most common immune defect in any type of chromosome 18 abnormality (Cody et al. 2015; Hasi-Zogaj et al. 2015). Distal hemizygosity of 18q22.3-q23 has been also shown to be associated with SIgAD (Dostal et al. 2007). However, normal serum IgA levels in our patient could be due the variable penetrance of this locus that has been estimated to be between 33% and 50% (Cody et al. 2015). Additionally, the fact that SIgAD also affects patients with 18p-, suggests that more than 1 single locus on chromosome 18 regulates IgA. Indeed, there is evidence that there is a common genetic basis for SIgAD and CVID as the occurrence of both diseases in the same family or progression of SIgAD to CVID has been observed (Aghamohammadi et al. 2008). The conversion of SIgAD to CVID has also been previously reported in a female patient with 18q-syndrome (Slyper and Pietryga 1997). Linkage analysis has failed to show any loci on chromosome 18 associated with SIgAD (Vorechovsky et al. 1999). Nonetheless, given the technical limitations at the time of those earlier studies, a re-assessment of patients using recently developed technologies such as arrays or next generation sequencing might help to discover novels gene defects or genomic variations implicated in SIgAD or CVID.
In our patient, both IgM and IgG serum levels were low and treatment with immunoglobulin was associated with reduction in the frequency of infections, elimination of septic episodes, and improved weight gain. She failed to develop a protective response to unconjugated pneumococcal capsule antigens despite adequate post-vaccine responses to tetanus and diphtheria, suggesting the diagnosis of specific antibody deficiency. The lack of specific polysaccharide antibodies has been previously reported in a patient with 18p- who only responded once immunized with a conjugated vaccine (Browning 2010). Meanwhile, a recent report of a patient with ring chromosome 18 depicted a different vaccine response: while all immunoglobulin isotypes, including IgA, were low, he had a good responses to unconjugated pneumococcal vaccination and booster doses of diphtheria and tetanus vaccines (Calvo Campoverde et al. 2016). Therefore, it is likely that, at least for protein vaccines, a booster dose can augment the specific antibody response in patients with chromosome 18 abnormalities and the use of conjugate vaccines should be prioritized.
Impaired homeostasis of memory T and B cells has been documented in CVID patients with autoimmunity (van de Ven and Warnatz 2015). We observed decreased CD19+CD27+ switched memory B cells and CD45RO+ memory T cell populations, as has been shown in 1 other patient with ring chromosome 18 (Calvo Campoverde et al. 2016). CVID patients with reduced number of CD45RO+ T cells might be affected with severe viral infections (Narula et al. 2007).
Association of primary antibody deficiencies and autoimmunity has been well characterized (van de Ven and Warnatz 2015). In addition to humoral immunodeficiency, patients with chromosome 18 abnormalities have been affected with rheumatoid arthritis, lupus, thyroiditis, vitiligo and type I diabetes mellitus (Dacou-Voutetakis et al. 1999; Hasi-Zogaj et al. 2015). In a patient with a terminal deletion of 18q (18q21.32-q23) and low serum IgA and IgG4 levels, autoimmune thyroiditis and type 1 diabetes mellitus, regulatory T cell (Treg) counts were low (Hogendorf et al. 2016). Our patient has severe polyarthritis, uveitis, and hypothyroidism. Unfortunately, we do not have data on Tregs in our patient.
The lack of efficient B cell and T cell interplay has been proposed to affect the development of memory T cells (Martini et al. 2011). Our understanding of the genetics of B cell deficiencies over the past decade has largely advanced. None of those genes is located on chromosome 18. In fact, patients with proximal interstitial deletions of 18q do not show any abnormal immune phenotype (Kato et al. 2010; Imataka et al. 2015). Therefore, it is probable that the candidate genes for antibody defects on 18q are located distally. In fact, homozygous pathogenic variants of Mucosa-Associated Lymphoid Tissue Lymphoma Translocation 1 gene (MALT1), that resides on 18q21.32, has been reported in patients with both CVID phenotypes with reduced switched memory B cells and autoimmunity with decreased Foxp3+ T cells (McKinnon et al. 2014; Charbit-Henrion et al. 2017). To examine if our patient’s immune dysregulation was due to a gene variant on the regions of deletion, including MALT1, and to exclude other known monogenic causes of immunodeficiency and autoimmunity, we performed WGS. However, we could not find any variant that could explain the immunological phenotype, nor could we find any pathogenic variant, including CNVs, of MALT1. To our knowledge, MALT1 haploinsufficiency has not been reported in humans as yet.
Indeed, few genes on chromosome 18 have been predicted to be haploinsufficient (Cody et al. 2009), among which only TCF4 falls into the deleted region in our patient. Heterozygous mutations of TCF4, however, are associated with Pitt-Hopkins syndrome with no well-established immunological presentation. We ranked the genes on the deleted regions, using the probability of being loss-of-function (LoF) intolerant (pLI), described by the Exome Aggregation Consortium (ExAC; exac.broadinstitute.org) (Lek et al. 2016). Genes with pLI ≥ 0.9 are considered as an extremely LoF intolerant, meaning haploinsufficient.
We reviewed published studies in Pubmed.com and further shortlisted genes of interest based on their possible involvement in immunological processes (Table 5). Interestingly, both TCF4 and MALT1 ranked on the top of the list (Table 5). This observation suggests that the phenotypical consequence of genetic variants of those genes might not have been recognized, including possible digenic conditions.
Table 5:
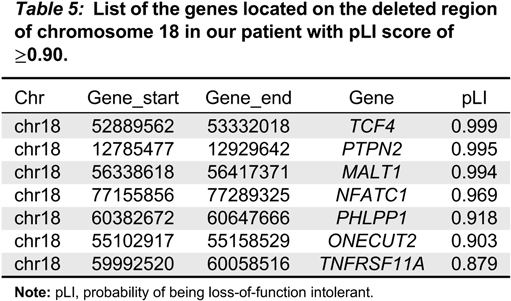
Note: pLI, probability of being loss-of-function intolerant.
We also examined other genes in the deleted region, determined from the literature, that could contribute to the lack of memory cells and SIgAD. PTPN2 is a negative regulator of Jak1 and Jak3 and deficient lymphocytes show increased STAT1 and STAT5 signaling (Simoncic et al. 2002). Ptpn2−/− mice show impaired B cell lymphopoiesis, as well as impaired T and B cell response to mitogens. However, T cell development in the thymus is not affected (You-Ten et al. 1997). On the other hand, PTPN2 expression is enhanced in B cell lymphomas (Lu et al. 2007). Importantly, it has been shown that early maturation of B cells in the bone marrow of Ptnp2−/− mice was blocked due to enhanced IFNγ-STAT1 signaling (Bourdeau et al. 2007). STAT1 activation is known to be upregulated in autoimmune disorders (Domeier et al. 2016). Also, gain-of-function (GOF) mutations of STAT1 have been shown in patients with CVID and autoimmunity (Al Rushood et al. 2013; Toubiana et al. 2016). Given the direct effect of PTPN2 on STAT1 phosphorylation (ten Hoeve et al. 2002), loss of PTPN2 might contribute to a similar phenotype as of STAT1 GOF mutations.
TNFRSF11A, also known as Receptor Activator of Nuclear Factor Kappa-B (RANK), encodes the receptor for RANK ligand (RANKL) that together make the master signaling pathway for osteoclast differentiation. Patients with Autosomal-Recessive Osteoporosis (ARO) are affected by homozygous mutations of RANK and RANKL. Additionally, these patients are unable to produce antibodies in response to tetanus vaccination (Guerrini et al. 2008). Interestingly, patients with homozygous mutations of TNFRSF11A (RANK), but not TNFSF11 (RANKL) show decreased switched memory B cells (IgD−CD27+), but did not show T cell abnormalities (Guerrini et al. 2008).
PHLPP1 is a phosphatase that in parallel with PTEN regulates the PI3K/AKT pathway (Chen et al. 2016). Heterozygous mutations of PTEN cause Hamartoma Tumor Syndromes (PHTS). Patients with PHTS have defective antibody responses and autoimmune manifestations (Driessen et al. 2016). While absolute numbers of transitional peripheral B cells are elevated in these patients, the CD27+ memory B cell population is decreased. Moreover, class switch recombination and somatic hypermutation are impaired in PHTS patients (Driessen et al. 2016) as well as in PTEN deficient mice (Suzuki et al. 2003). The effect of loss of PTEN on B cells has been attributed to increased PI3K/AKT signaling. In fact, in patients with dominant GOF mutations of P110δ (a subunit of PI3K electively expressed in lymphocytes) show impaired class switch recombination, defective antibody responses and increased transitional B cells (Angulo et al. 2013). Notably, although loss of PTEN in humans does not affect Treg development, inhibitors of PHLPP1 block in vitro Treg differentiation (Chen et al. 2016). Collectively, absence of PHLPP1 in patients with 18q- could putatively replicate the immunological phenotype of PTEN deficiency in association with defective Treg responses. We found a deep intronic 4 kb deletion in intron 8 of PHLPP1 in our patient. However, this deletion is very unlikely to be deleterious as it is quite far from flanking splice sites. Further functional studies will be required to assess whether haploinsufficiency of PHLPP1 is could lead to this immunological phenotype.
NFATC1, encoding the nuclear factor of activated T cell c1 (NFATc1), is a member of NFAT family of transcriptional factors that are regulated via Ca2+/Calcineurin (Medyouf and Ghysdael 2008). Based on animal models, it has been suggested that NFATc1 deficiency might cause immunodeficiency in human patients with 18q- (Li et al. 1995). Loss of Nfatc1 in mice affects development and survival of both peritoneal and splenic B1-a cells (Berland and Wortis 2003). Moreover, even though NFATc1 does not affect the development and maturation of B cells, it plays a critical role in their function and fate. It has been shown that Nfact1−/− B cells in mice show impaired proliferation and survival in response to BCR stimulation, Ig class switching and can suppress T cell activation (Bhattacharyya et al. 2011). These findings were in association with impaired Ca2+ influx resembling defective BCR signaling (Bhattacharyya et al. 2011). On the other hand, loss of NFATc1 in T cells impaired homing of follicular regulatory T cells in B cell follicles via transactivation of CXCR5. This was associated with an exacerbated lupus-like phenotype in mice (Bhattacharyya et al. 2011).
Conclusion
The association of hypogammaglobulinemia and abnormal chromosome 18 was recognized over 50 years ago (Feingold and Schwartz 1968). However, despite the advancement of our understanding of the genetics of the immune system, the responsible gene(s) for this phenotype are not yet known. In this case, in addition to low serum immunoglobulin levels, we found that both T and B memory cells and specific polysaccharide antibody responses were defective in our patient with ring chromosome 18. These data suggest that although the underlying defective mechanisms are not yet elucidated, it might be beneficial to attempt booster doses of protein or conjugate vaccines to patients with chromosome 18 abnormalities and defective humoral immunity. Further investigations on patients with chromosome 18 aberrations using new technologies could help discover novel genes involved in primary immunodeficiency and autoimmunity.
REFERENCES
Aghamohammadi A., Mohammadi J., Parvaneh N., Rezaei N., Moin M., Espanol T., and Hammarstrom L. 2008. Progression of selective IgA deficiency to common variable immunodeficiency. Int. Arch. Allergy Immunol. 147(2):87–92.
Al Rushood M., McCusker C., Mazer B., Alizadehfar R., Grimbacher B., Depner M., and Ben-Shoshan M. 2013. Autosomal dominant cases of chronic mucocutaneous candidiasis segregates with mutations of signal transducer and activator of transcription 1, but not of Toll-like receptor 3. J. Pediatr. 163(1):277–279.
Angulo I., Vadas O., Garcon F., Banham-Hall E., Plagnol V., Leahy T.R., Baxendale H., Coulter T., Curtis J., Wu C., Blake-Palmer K., Perisic O., Smyth D., Maes M., Fiddler C., Juss J., Cilliers D., Markelj G., Chandra A., Farmer G., Kielkowska A., Clark J., Kracker S., Debre M., Picard C., Pellier I., Jabado N., Morris J.A., Barcenas-Morales G., Fischer A., Stephens L., Hawkins P., Barrett J.C., Abinun M., Clatworthy M., Durandy A., Doffinger R., Chilvers E.R., Cant A.J., Kumararatne D., Okkenhaug K., Williams R.L., Condliffe A., and Nejentsev S. 2013. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. 342(866):866–871.
Benini R., Saint-Martin C., Shevell M.I., and Bernard G. 2012. Abnormal myelination in ring chromosome 18 syndrome. J. Child Neurol. 27(8):1042–1047.
Berland R. and Wortis H.H. 2003. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc. Natl. Acad. Sci. USA. 100(23):13459–13464.
Bhattacharyya S., Deb J., Patra A.K., Pham D.A.T., Chen W., Vaeth M., Berberich-Siebelt F., Klein-Hessling S., Lamperti E.D., Reifenberg K., Jellusova J., Schweizer A., Nitschke L., Leich E., Rosenwald A., Brunner C., Engelmann S., Bommhardt U., Avots A., Muller M.R., Kondo E., and Serfling E. 2011. NFATc1 affects mouse splenic B cell function by controlling the calcineurin–NFAT signaling network. J. Exp. Med. 208(4):823–839.
Bourdeau A., Dube N., Heinonen K.M., Theberge J.F., Doody K.M., and Tremblay M.L. 2007. TC-PTP-deficient bone marrow stromal cells fail to support normal B lymphopoiesis due to abnormal secretion of interferon-γ. Blood. 109(10):4220–4228.
Browning M.J. 2010. Specific polysaccharide antibody deficiency in chromosome 18p deletion syndrome and immunoglobulin A deficiency. J. Investig. Allergol. Clin. Immunol. 20(3):263–266.
Calvo Campoverde K., Gean E., Piquer Gibert M., Martinez Valdez L., Deya-Martinez A., Rojas Volquez M., Esteve-Sole A., Juan M., Plaza M.A., and Alsina L. 2016. Humoral deficiency in three paediatric patients with genetic diseases. Allergol. Immunopathol. 44(3):257–262.
Carter E., Heard P., Hasi M., Soileau B., Sebold C., Hale D.E., and Cody J.D. 2015. Ring 18 molecular assessment and clinical consequences. Am. J. Med. Genet., Part A. 167A(1):54–63.
Charbit-Henrion F., Jeverica A.K., Begue B., Markelj G., Parlato M., Avcin S.L., Callebaut I., Bras M., Parisot M., Jazbec J., Homan M., Ihan A., Rieux-Laucat F., Stolzenberg M.C., Ruemmele F.M., Avcin T., Cerf-Bensussan N., and GENIUS Group. 2017. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: A novel cause of IPEX-like syndrome. J. Pediatr. Gastroenterol. Nutr. 64(3):378–384.
Chen H.H., Handel N., Ngeow J., Muller J., Huhn M., Yang H.T., Heindl M., Berbers R.M., Hegazy A.N., Kionke J., Yehia L., Sack U., Blaser F., Rensing-Ehl A., Reifenberger J., Keith J., Travis S., Merkenschlager A., Kiess W., Wittekind C., Walker L., Ehl S., Aretz S., Dustin M.L., Eng C., Powrie F., and Uhlig H.H. 2016. Immune dysregulation in patients with PTEN hamartoma tumor syndrome: Analysis of FOXP3 regulatory T cells. J. Allergy Clin. Immunol. 139(2):607–620.e15.
Cody J.D., Carter E.M., Sebold C., Heard P.L., and Hale D.E. 2009. A gene dosage map of Chromosome 18: A map with clinical utility. Genet. Med. 11(11):778–782.
Cody J.D., Hasi M., Soileau B., Heard P., Carter E., Sebold C., O’Donnell L., Perry B., Stratton R.F., and Hale D.E. 2014. Establishing a reference group for distal 18q-: Clinical description and molecular basis. Hum. Genet. 133(2):199–209.
Cody J.D., Sebold C., Heard P., Carter E., Soileau B., Hasi-Zogaj M., Hill A., Rupert D., Perry B., O’Donnell L., Gelfond J., Lancaster J., Fox P.T., and Hale D.E. 2015. Consequences of chromsome18q deletions. Am. J. Med. Genet., Part C: Semin. Med. Genet. 169(3):265–280.
Conley M.E. 2009. Genetics of hypogammaglobulinemia: What do we really know? Curr. Opin. Immunol. 21(5):466–471.
Cunningham-Rundles C. 2012. Human B cell defects in perspective. Immunol. Res. 54(1–3):227–232.
Dacou-Voutetakis C., Sertedaki A., Maniatis-Christidis M., Sarri C., Karadima G., Petersen M.B., Xaidara A., Kanariou M., and Nicolaidou P. 1999. Insulin dependent diabetes mellitus (IDDM) and autoimmune thyroiditis in a boy with a ring chromosome 18: Additional evidence of autoimmunity or IDDM gene(s) on chromosome 18. J. Med. Genet. 36(2):156–158.
Domeier P.P., Chodisetti S.B., Soni C., Schell S.L., Elias M.J., Wong E.B., Cooper T.K., Kitamura D., and Rahman Z.S. 2016. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J. Exp. Med. 213(5):715–732.
Dostal A., Linnankivi T., Somer M., Kahkonen M., Litzman J., and Tienari P. 2007. Mapping susceptibility gene locus for IgA deficiency at del(18)(q22.3-q23); report of familial cryptic chromosome t(18q; 10p) translocations. Int. J. Immunogenet. 34(3):143–147.
Driessen G.J., IJspeert H., Wentink M., Yntema H.G., van Hagen P.M., van Strien A., Bucciol G., Cogulu O., Trip M., Nillesen W., Peeters E.A., Pico-Knijnenburg I., Barendregt B.H., Rizzi M., van Dongen J.J., Kutukculer N., and van der Burg M. 2016. Increased PI3K/Akt activity and deregulated humoral immune response in human PTEN deficiency. J. Allergy Clin. Immunol. 138(6):1744–1747.e5.
Faed M.J., Whyte R., Paterson C.R., McCathie M., and Robertson J. 1972. Deletion of the long arms of chromosome 18 (46,XX,18q-) associated with absence of IgA and hypothyroidism in an adult. J. Med. Genet. 9(1):102–105.
Feingold M. and Schwartz R.S. 1968. IgA and partial deletions of chromosome 18. Lancet. 292(7577):1086.
Fischer P., Golob E., Friedrich F., Kunze-Muhl E., Doleschel W., and Aichmair H. 1970. Autosomal deletion syndrome 46,XX,18p-: A new case report with absence of IgA in serum. J. Med. Genet. 7(1):91–98.
Guerrini M.M., Sobacchi C., Cassani B., Abinun M., Kilic S.S., Pangrazio A., Moratto D., Mazzolari E., Clayton-Smith J., Orchard P., Coxon F.P., Helfrich M.H., Crockett J.C., Mellis D., Vellodi A., Tezcan I., Notarangelo L.D., Rogers M.J., Vezzoni P., Villa A., and Frattini A. 2008. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am. J. Hum. Genet. 83(1):64–76.
Guilherme R.S., Meloni V.F., Kim C.A., Pellegrino R., Takeno S.S., Spinner N.B., Conlin L.K., Christofolini D.M., Kulikowski L.D., and Melaragno M.I. 2011. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med. Genet. 12:171.
Hasi-Zogaj M., Sebold C., Heard P., Carter E., Soileau B., Hill A., Rupert D., Perry B., Atkinson S., O’Donnell L., Gelfond J., Lancaster J., Fox P.T., Hale D.E., and Cody J.D. 2015. A review of 18p deletions. Am. J. Med. Genet., Part C: Semin. Med. Genet. 169(3):251–264.
Hogendorf A., Lipska-Zietkiewicz B.S., Szadkowska A., Borowiec M., Koczkowska M., Trzonkowski P., Drozdz I., Wyka K., Limon J., and Mlynarski W. 2016. Chromosome 18q deletion syndrome with autoimmune diabetes mellitus: Putative genomic loci for autoimmunity and immunodeficiency. Pediatr. Diabetes. 17(2):153–159.
Imataka G., Ohwada Y., Shimura N., Yoshihara S., and Arisaka O. 2015. Del(18)(q12.2q21.1) syndrome: A case report and clinical review of the literature. Eur. Rev. Med. Pharmacol. Sci. 19(17):3241–3245.
Kato Z., Morimoto W., Kimura T., Matsushima A., and Kondo N. 2010. Interstitial deletion of 18q: Comparative genomic hybridization array analysis of 46, XX,del(18)(q21.2.q21.33). Birth Defects Res., Part A: Clin. Mol. Teratol. 88(2):132–135.
Lejtenyi D. and Mazer B. 2008. Consistency of protective antibody levels across lots of intravenous immunoglobulin preparations. J. Allergy Clin. Immunol. 121(1):254–255.
Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Tukiainen T., Birnbaum D.P., Kosmicki J.A., Duncan L.E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D.N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M.I., Moonshine A.L., Natarajan P., Orozco L., Peloso G.M., Poplin R., Rivas M.A., Ruano-Rubio V., Rose S.A., Ruderfer D.M., Shakir K., Stenson P.D., Stevens C., Thomas B.P., Tiao G., Tusie-Luna M.T., Weisburd B., Won H.H., Yu D., Altshuler D.M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J.C., Gabriel S.B., Getz G., Glatt S.J., Hultman C.M., Kathiresan S., Laakso M., McCarroll S., McCarthy M.I., McGovern D., McPherson R., Neale B.M., Palotie A., Purcell S.M., Saleheen D., Scharf J.M., Sklar P., Sullivan P.F., Tuomilehto J., Tsuang M.T., Watkins H.C., Wilson J.G., Daly M.J., MacArthur D.G., and Exome Aggregation Consortium. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536(7616):285–291.
Li X., Ho S.N., Luna J., Giacalone J., Thomas D.J., Timmerman L.A., Crabtree G.R., and Francke U. 1995. Cloning and chromosomal localization of the human and murine genes for the T-cell transcription factors NFATc and NFATp. Cytogenet. Cell Genet. 68(3–4):185–191.
Lu X., Chen J., Sasmono R.T., Hsi E.D., Sarosiek K.A., Tiganis T., and Lossos I.S. 2007. T-cell protein tyrosine phosphatase, distinctively expressed in activated-B-cell-like diffuse large B-cell lymphomas, is the nuclear phosphatase of STAT6. Mol. Cell. Biol. 27(6):2166–2179.
Martini H., Enright V., Perro M., Workman S., Birmelin J., Giorda E., Quinti I., Lougaris V., Baronio M., Warnatz K., and Grimbacher B. 2011. Importance of B cell co-stimulation in CD4(+) T cell differentiation: X-linked agammaglobulinaemia, a human model. Clin. Exp. Immunol. 164(3):381–387.
McKinnon M.L., Rozmus J., Fung S.Y., Hirschfeld A.F., Del Bel K.L., Thomas L., Marr N., Martin S.D., Marwaha A.K., Priatel J.J., Tan R., Senger C., Tsang A., Prendiville J., Junker A.K., Seear M., Schultz K.R., Sly L.M., Holt R.A., Patel M.S., Friedman J.M., and Turvey S.E. 2014. Combined immunodeficiency associated with homozygous MALT1 mutations. J. Allergy Clin. Immunol. 133(5):1458–1462.e7.
Medyouf H. and Ghysdael J. 2008. The calcineurin/NFAT signaling pathway: A novel therapeutic target in leukemia and solid tumors. Cell Cycle. 7(3):297–303.
Michaels D.L., Go S., Humbert J.R., Dubois R.S., Stewart J.M., and Ellis E.F. 1971. Intestinal nodular lymphoid hyperplasia, hypogammaglobulinemia, and hematologic abnormalities in a child with a ring 18 chromosome. J. Pediatr. 79(1):80–88.
Monlong J., Girard S.L., Meloche C., Cadieux-Dion M., Andrade D.M., Lafreniere R.G., Gravel M., Spiegelman D., Dionne-Laporte A., Boelman C., Hamdan F.F., Michaud J.L., Rouleau G., Minassian B.A., Bourque G., and Cossette P. 2018. Global characterization of copy number variants in epilepsy patients from whole genome sequencing. PLoS Genet. 14(4):e1007285.
Narula S., LaRosa D.F., Kamoun M., Dalmau J., and Levinson A.I. 2007. Progressive multifocal leukoencephalopathy in a patient with common variable immunodeficiency and abnormal CD8+ T-cell subset distribution. Ann. Allergy Asthma Immunol. 98(5):483–489.
Schatorje E., van der Flier M., Seppanen M., Browning M., Morsheimer M., Henriet S., Neves J.F., Vinh D.C., Alsina L., Grumach A., Soler-Palacin P., Boyce T., Celmeli F., Goudouris E., Hayman G., Herriot R., Forster-Waldl E., Seidel M., Simons A., and de Vries E. 2016. Primary immunodeficiency associated with chromosomal aberration—An ESID survey. Orphanet J. Rare Dis. 11(1):110.
Schinzel A., Schmid W., Luscher U., Nater M., Brook C., and Steinmann B. 1974. Structural aberrations of chromosome 18. I. The 18p-syndrome. Arch. Genet. 47(1):1–15.
Simoncic P.D., Lee-Loy A., Barber D.L., Tremblay M.L., and McGlade C.J. 2002. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr. Biol. 12(6):446–453.
Slyper A.H. and Pietryga D. 1997. Conversion of selective IgA deficiency to common variable immunodeficiency in an adolescent female with 18q deletion syndrome. Eur. J. Pediatr. 156(2):155–156.
Suzuki A., Kaisho T., Ohishi M., Tsukio-Yamaguchi M., Tsubata T., Koni P.A., Sasaki T., Mak T.W., and Nakano T. 2003. Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J. Exp. Med. 197(5):657–667.
ten Hoeve J., de Jesus Ibarra-Sanchez M., Fu Y., Zhu W., Tremblay M., David M., and Shuai K. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 22(16):5662–5668.
Toubiana J., Okada S., Hiller J., Oleastro M., Gomez M.L., Becerra J.C.A., Ouachee-Chardin M., Fouyssac F., Girisha K.M., Etzioni A., Van Montfrans J., Camcioglu Y., Kerns L.A., Belohradsky B., Blanche S., Bousfiha A., Rodriguez-Gallego C., Meyts I., Kisand K., Reichenbach J., Renner E.D., Rosenzweig S., Grimbacher B., van de Veerdonk F.L., Traidl-Hoffmann C., Picard C., Marodi L., Morio T., Kobayashi M., Lilic D., Milner J.D., Holland S., Casanova J.L., Puel A., and on behalf of the International STAT1 Gain-of-Function Study Group. 2016. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 127(25):3154–3164.
van de Ven A.A. and Warnatz K. 2015. The autoimmune conundrum in common variable immunodeficiency disorders. Curr. Opin. Allergy Clin. Immunol. 15(6):514–524.
Vorechovsky I., Blennow E., Nordenskjold M., Webster A.D., and Hammarstrom L. 1999. A putative susceptibility locus on chromosome 18 is not a major contributor to human selective IgA deficiency: Evidence from meiotic mapping of 83 multiple-case families. J. Immunol. 163(4):2236–2242.
You-Ten K.E., Muise E.S., Itie A., Michaliszyn E., Wagner J., Jothy S., Lapp W.S., and Tremblay M.L. 1997. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 186(5):683–693.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 7 • Number 1 • March 2020
Pages: 25 - 36
History
Received: 17 August 2019
Accepted: 12 November 2019
Accepted manuscript online: 20 December 2019
Copyright
© 2020.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MehdiYeganeh, TallalBasha, Lina SobhiAbdrabo, Sophie RanWang, JoëlLafond-Lapalme, Jean-BaptisteRivière, DuncanLejtenyi, David S.Rosenblatt, ChristineMcCusker, RezaAlizadehfar, and Bruce D.Mazer. 2020. Primary antibody deficiency associated with ring chromosome 18. LymphoSign Journal.
7(1): 25-36. https://doi.org/10.14785/lymphosign-2019-0013
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
