Use of induced pluripotent stem cells to investigate the effects of purine nucleoside phosphorylase deficiency on neuronal development
Abstract
Background: Inherited defects in the function of the purine nucleoside phosphorylase (PNP) enzyme can cause severe T cell immune deficiency and early death from infection, autoimmunity, or malignancy. In addition, more than 50% of patients suffer diverse non-infectious neurological complications. However the cause for the neurological abnormalities are not known.
Objectives: Differentiate induced pluripotent stem cells (iPSC) from PNP-deficient patients into neuronal cells to better understand the effects of impaired purine metabolism on neuronal development.
Methods: Sendai virus was used to generate pluripotent stem cells from PNP-deficient and healthy control lymphoblastoid cells. Cells were differentiated into neuronal cells through the formation of embryoid bodies.
Results: After demonstration of pluripotency, normal karyotype, and retention of the PNP deficiency state, iPSC were differentiated into neuronal cells. PNP-deficient neuronal cells had reduced soma and nuclei size in comparison to cells derived from healthy controls. Spontaneous apoptosis, determined by Caspase-3 expression, was increased in PNP-deficient cells.
Conclusions: iPSC from PNP-deficient patients can be differentiated into neuronal cells, thereby providing an important tool to study the effects of impaired purine metabolism on neuronal development and potential treatments.
Statement of novelty: We report here the first generation and use of neuronal cells derived from induced pluripotent stem cells to model human PNP deficiency, thereby providing an important tool for better understanding and management of this condition.
Introduction
Purine nucleoside phosphorylase (PNP) is an important enzyme in the phosphorylation of guanosine and deoxyguanosine to inosine and hypoxanthine, which can eventually be excreted as uric acid (Figure 1). Alternatively, deoxyguanosine can be phosphorylated by deoxyguanosine kinase to dGTP. Inherited defects in PNP, resulting in <5% enzyme activity, cause an accumulation of PNP substrates. There is also depletion of PNP products, often leading to a reduction in hypoxanthine and uric acid (Cohen et al. 2000). The impaired purine metabolism leads to enhanced thymocyte apoptosis, reduced secretion of cytokines by immune cells, and impaired proliferation of peripheral T cells in response to mitogens and allogeneic cells (Arpaia et al. 2000; Grunebaum et al. 2004; Yu et al. 2009; Papinazath et al. 2011). The T cell dysfunction results in increased susceptibility to infections, autoimmunity, and malignancy (Watson et al. 1981; Delicou et al. 2007). In some patients, defects in NK- and B-lymphocytes were also reported, further increasing patients’ risk for infections (Somech et al. 2013). PNP is ubiquitously expressed, suggesting that PNP deficiency might also affect non-lymphoid tissues. Indeed, dysplastic bone marrow with increased sensitivity of bone marrow cells to irradiation has previously been reported (Dror et al. 2004). In addition, more than 50% of patients with PNP deficiency exhibit diverse non-infectious neurological dysfunction that often precede the immunological abnormalities, and persist even after correction of the immune deficiency (Baguette et al. 2002; Tabarki et al. 2003; Ozkinay et al. 2007; Grunebaum et al. 2013).
Figure 1:
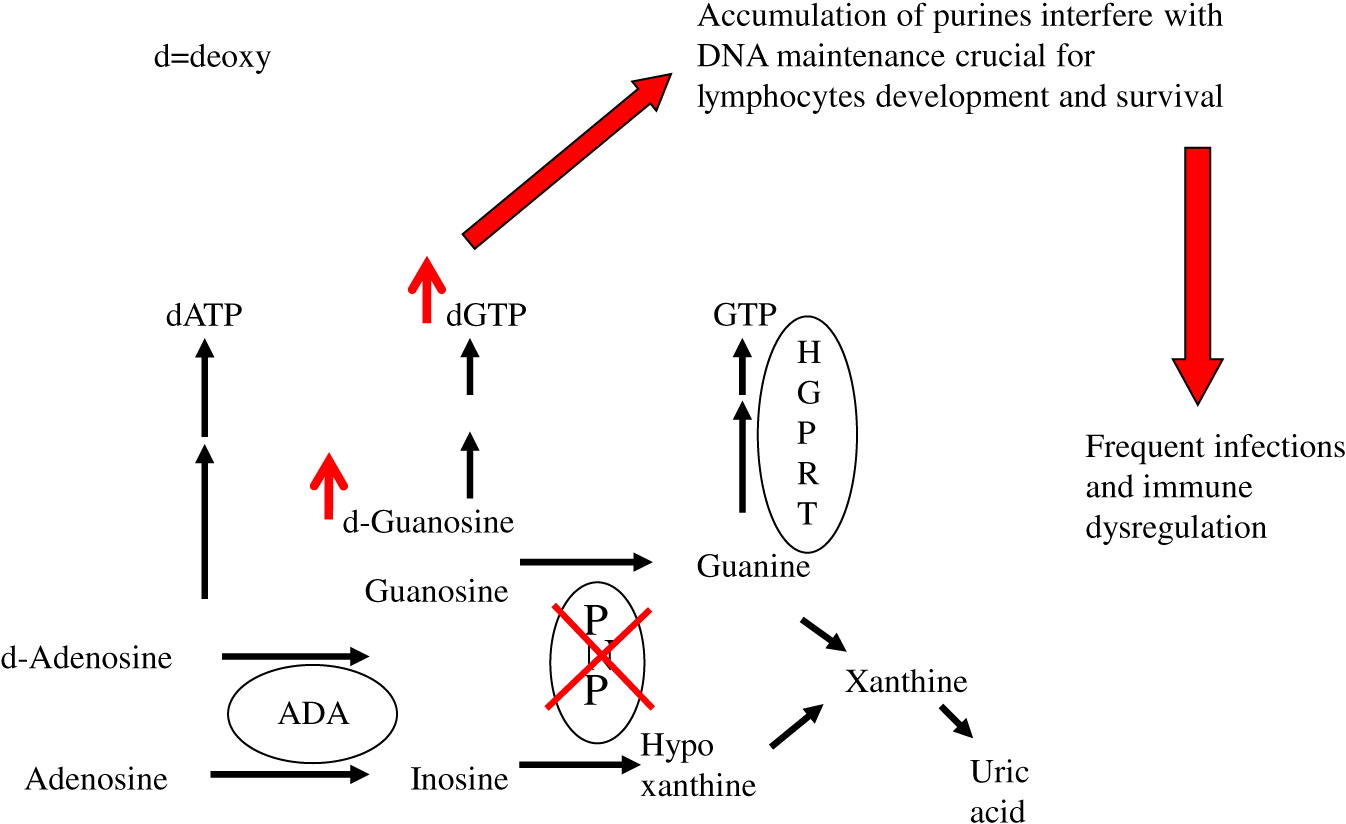
Limited access to tissues from patients and ethical considerations have impeded the ability to study mechanisms responsible for the abnormalities observed in PNP-deficient patients. A PNP-deficient (PNP−/−) mouse model that recapitulated many of the human manifestations has allowed overcoming some of these limitations (Arpaia et al. 2000). PNP−/− mice demonstrated decreased ability to remain on a revolving rota-rod, an assay used to assess ataxia in mice, as well as smaller cerebellum, reduced number of cerebellar Purkinje cells and increased apoptosis of neurons, in comparison to healthy PNP-proficient littermate mice (Mansouri et al. 2012). Moreover, repeated injections with PNP fused to the HIV TAT protein transduction domain (Toro and Grunebaum 2006) corrected the PNP deficiency and prevented the occurrence of these abnormalities (Mansouri et al. 2012). However, the precise mechanism for the neuronal apoptosis was not determined, nor was it clear whether the immune abnormalities observed in the PNP−/− mice contributed to the phenotype.
Induced pluripotent stem cells (iPSC) have recently been used to better understand the pathogenesis of diverse neurological disorders (Barral and Kurian 2016). Using lymphocytes, fibroblasts, skin cells and other sources, which can be de-differentiated into iPSC and then matured into specific lineages, iPSC have been shown to be a robust tool in generating unlimited amounts of cells that simulate many stages of human neuronal development (Mertens et al. 2016).
Accordingly, we hypothesized that studying neurons cells differentiated from the iPSC of PNP-deficient patients would enable better appreciation of the role that perturbed PNP metabolism has on neurons, as well as treatments for this condition. Here we present preliminary results demonstrating the utility of such cells in evaluating the effects of PNP deficiency on neuron cell development.
Methods
EBV-transformed lymphocytes from PNP-deficient patient and healthy controls were used to generate iPSC lines by the Canadian Center for Regenerative Medicine (Toronto, ON) with Sendai viruses, as described previously (Patel and Yang 2010). PNP activity of the cells was assessed by measuring conversion of [8-14C]inosine (50 mCi/mmol; Moravek Biochemicals, Brea, CA, USA) to hypoxanthine using cellulose TLC, as described previously (Toro et al. 2006).
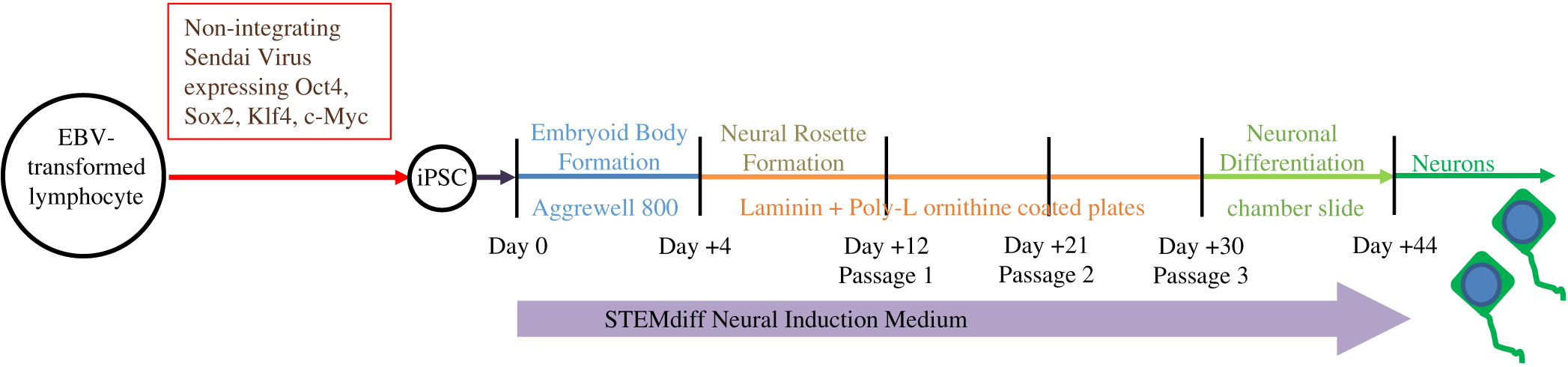
As described in Figure 2, iPSC were differentiated through the formation of embryoid bodies by dissociating iPSC and plating 2 × 106 cells/well in Aggrewell 800 plates (STEMCELL Technologies, Vancouver, BC). After 4 days, embryoid bodies were dissociated and plated onto Laminin and Poly-l-ornithine coated 6-well plates for an additional 7 days. Following formation of neural rosettes, rosettes were detached using STEMdiff Neural Rosette Selection Reagent (STEMCELL) and plated onto Laminin and Poly-l-ornithine coated plates at a minimum density of 2 × 106 cells/well. The neural progenitor cells were then cultured to passage 3 and differentiated into neurons by plating 1 × 105 cells/well into chamber-slides. STEMdiff Neural Induction Medium (STEMCELL), which is serum free and devoid of PNP activity, was utilized throughout the entire differentiation.
Figure 2:
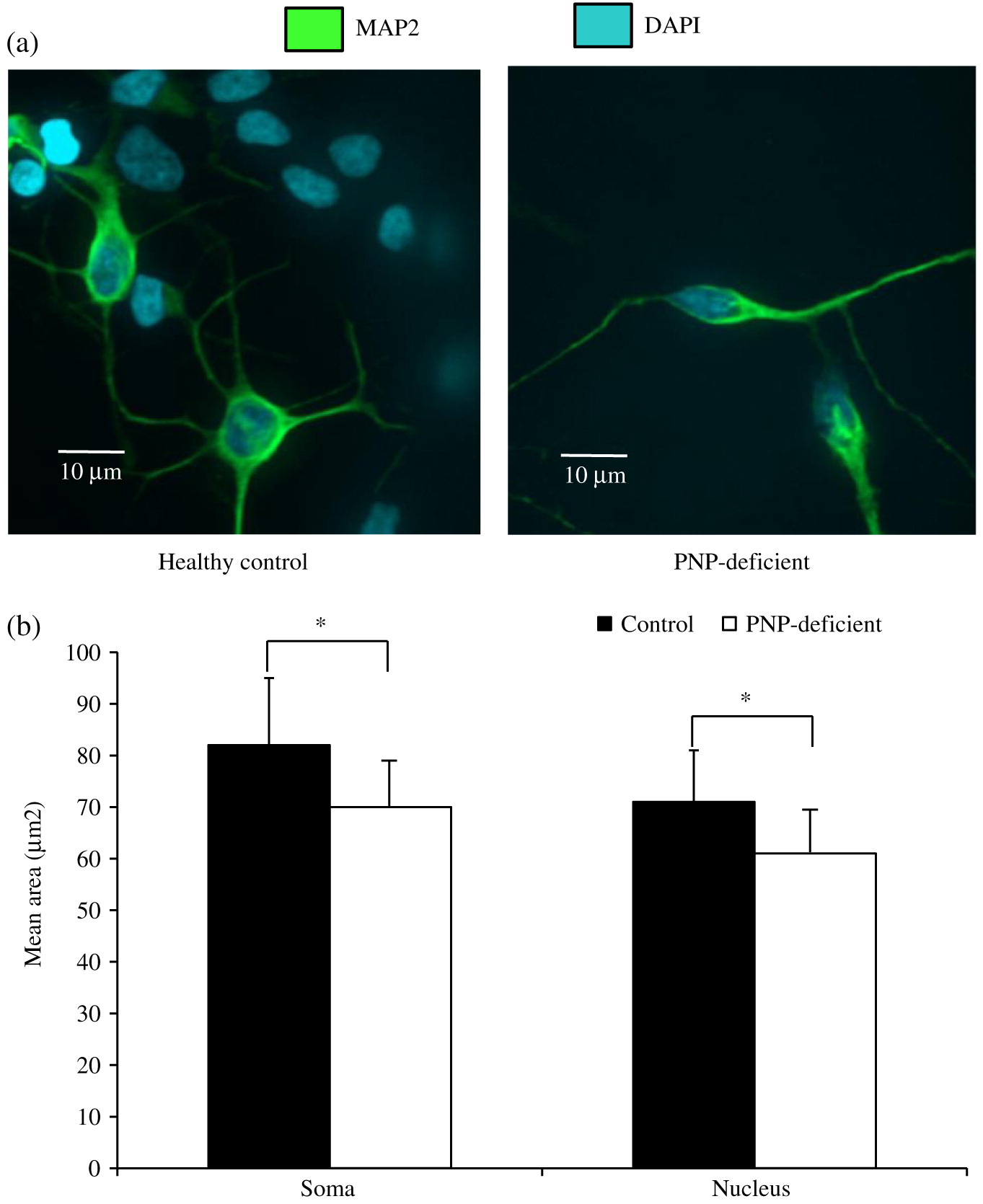
Neuronal cell soma and nuclei were stained with anti-MAP2 antibody (Abcam, Cambridge, MA, USA) and DAPI (ThermoFisher, Waltham, MA, USA), respectively. Soma and nuclei size analysis was performed using Improvision Volocity software (PerkinElmer, Woodbridge, ON) on images from more than 150 randomly selected fields from 30 cover slips over 3 independent biological replicates of neuronal differentiation per iPSC. Team members unaware to the PNP status of the cells performed these measurements. Apoptosis was detected by expression of cleaved Caspase-3 (Abcam) among cells mounted on slides. Statistical significance was determined using unpaired equal variance t test.
Results
Pluripotency and enzyme deficiency maintained in PNP-deficient iPSC
PNP-deficient EBV-transformed lymphocytes were de-differentiated into iPSC at similar efficacy as healthy controls. Pluripotency associated proteins SSEA4, OCT4, and NANOG were expressed by PNP-deficient iPSC, and these cells maintained normal karyotype and pluripotency potential similar to healthy control cells, indicating that PNP deficiency did not compromise generation of iPSC. Importantly, iPSC derived from the patients’ cells as well as neuronal cells generated from the iPSC maintained near absence of PNP enzyme activity (<1%), indicating their suitability to assess the effects of the PNP deficiency.
PNP-deficient iPSC have reduced cell and nucleus size
Confocal microscopy demonstrated that both PNP-deficient and proficient iPSC-derived neuronal cells, had morphological features similar to human neurons with dendrites projecting from the soma, and axons extending from axon hillocks (Figure 3a). As expected, these cells also expressed the neuron specific cytoskeletal protein microtubule associated protein (MAP)-2. However, neuronal cells differentiated from human PNP-deficient iPSC had reduced soma and nuclear sizes. Analysis of over 150 PNP-deficient cells showed significantly (p < 0.01) smaller soma and nuclei areas compared to cells derived from healthy controls (Figure 3b).
Figure 3:
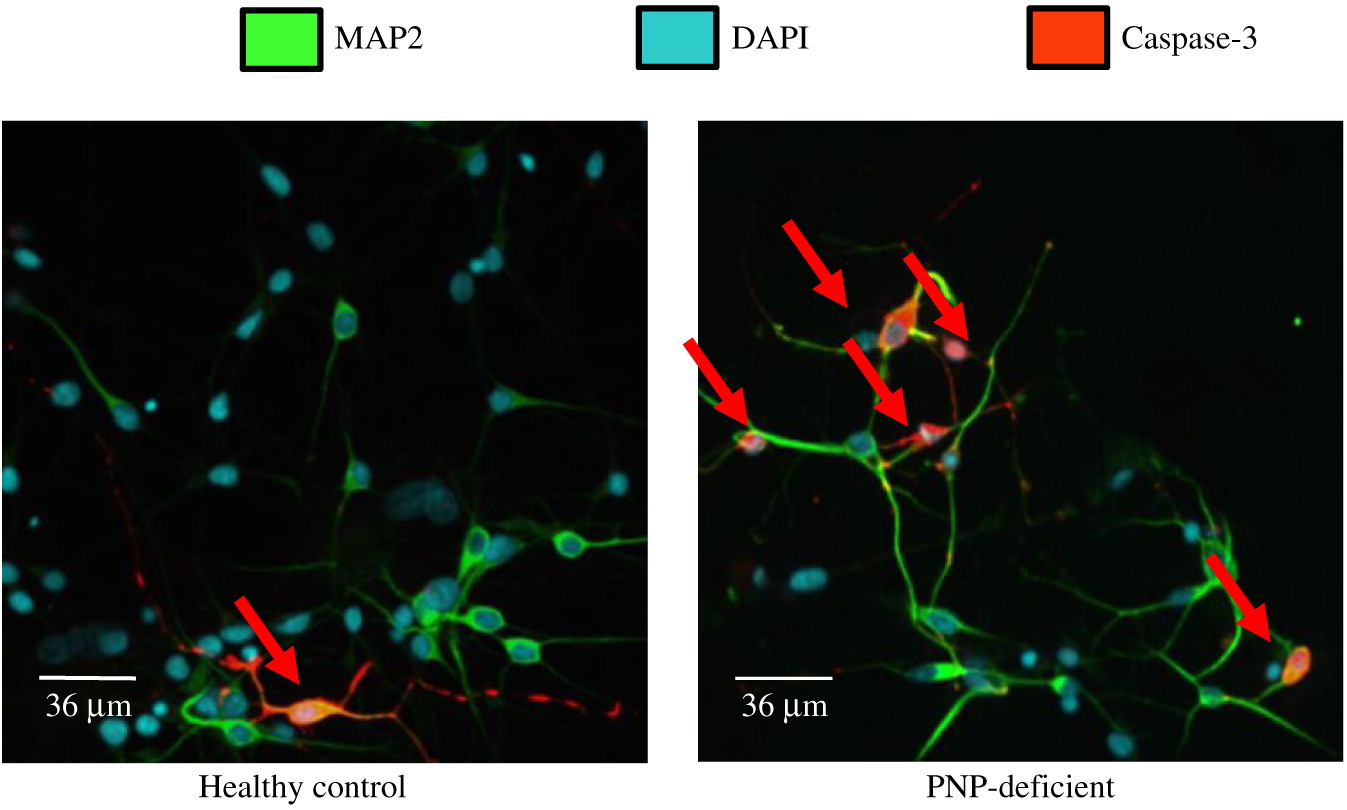
PNP-deficient iPSC demonstrate enhanced apoptosis
Detection of cleaved Caspase-3 was used to determine apoptosis in iPSC derived neuronal cells (Figure 4). Cleaved caspase-3 was expressed on 7.7% ± 4.0% of PNP-deficient neuronal cells, which was significantly (p < 0.01) higher than the 3.6% ± 3.9% PNP-proficient healthy control cells expressing cleaved Caspase-3, demonstrating increased spontaneous apoptosis of PNP-deficient derived neuronal cells.
Figure 4:
Discussion
Studying the neurological effects of impaired PNP function has been challenging as many patients suffer from infections, autoimmunity, and neurological abnormalities, which might impact their growth and development with secondary effects on brain development. Moreover, obtaining brain tissue is ethically unacceptable unless patients are undergoing evaluations for extensive neurological damage or succumb to their disease. As an alternative, we and other researchers have used animal models to better understand the effects of inherited purine defects on brain development, although differences in brain formation and function between mice and human limit the ability to draw conclusions relevant to humans (Jinnah et al. 1990; Mansouri et al. 2012; Sauer et al. 2017). Accordingly, the availability of patient-derived iPSC, which can be continuously propagated and directly differentiated to simulate human mature neuronal cells is an important tool. Indeed, in recent years the number of inherited neurological diseases that are modeled by iPSC has grown remarkably (Andrade et al. 2012; Chailangkarn et al. 2016; Allende et al. 2018).
Some barriers to reprogramming of differentiated cells into iPSC have been identified, such as senescence, expression of growth suppressor genes or increased sensitivity of the cells’ DNA to various noxious damage (Andrade et al. 2012). Although PNP is a house keeping enzyme important also for DNA and RNA formation and repair, PNP seems to be dispensable for generating pluripotent stem cells. This finding is in agreement with the development of PNP−/− mice embryonic stem cells, evident by the birth of PNP−/− pups from mating PNP−/− female and male mice (E. Grunebaum, unpublished data).
Our results show that while PNP deficiency does not prevent generation of neuronal cells, the PNP-deficient cells have impaired morphology and survival. The relevance of PNP-deficient iPSC derived neuronal cells’ small soma and nuclei size to the neurological and histological abnormalities observed in PNP-deficient patients and mice is still not clear. Although needed to be replicated in neuronal cells derived from other PNP-deficient patients, our findings are similar to those reported among iPSC derived from patients with Rett syndrome, a severe neurological disease caused by MECP2 mutations (Cheung et al. 2011), supporting the validity of our data. Moreover, it will be important to determine whether PNP deficiency also distorts the morphology of iPSC that can now be developed using new techniques into cerebellum- and purkinje cells (Watson et al. 2018), as observed in PNP−/− mice (Mansouri et al. 2012).
Interestingly, we found increased apoptosis in PNP-deficient neuronal cells, similar to the enhanced cells’ death already identified in PNP−/− mice thymocytes and cerebellar Purkinje cells (Papinazath et al. 2011; Mansouri et al. 2012). Although the spontaneous apoptosis of PNP-deficient neuronal cells was only about 2 fold higher than in normal cells, this rate is not much different than the 3–4 fold apoptosis increase in Cockayne- and Williams-syndrome iPSC derived cells, respectively (Andrade et al. 2012; Chailangkarn et al. 2016). Hence our data suggests that in progressive neurological diseases of childhood, such as PNP deficiency, even a small increase in apoptosis might be clinically relevant. The precise mechanism(s) leading to the apoptosis in PNP-deficient neuronal cells has not been established yet, however we have previously shown that the apoptosis in PNP deficiency is typically initiated in the mitochondria, and can occur in response to different conditions. These include exposure to deoxyguanosine, dexamethasone, or irradiation (Dror et al. 2004; Papinazath et al. 2011). Hence, it will be important to assess susceptibility of PNP-deficient neuronal cells to these factors as they might be generated or utilized when treating PNP-deficient patients for infections, autoimmunity, malignancy, or performing hematopoietic stem cell (HSC) transplantations. Better appreciation of the causes of PNP-deficient neuronal cell death might prompt avoidance of such compromising situations.
The availability of iPSC derived neuronal cells will also provide an opportunity to assess the effects of different treatment options on the neurological abnormalities associated with PNP deficiency. Transfusions of normal red blood cells rich in PNP (Rich et al. 1980), transplants with normal allogeneic (Yeates et al. 2017) or gene corrected autologous (Liao et al. 2008) HSC transplantations or frequent injections of native (Toro and Grunebaum 2006) or PEGylated (E. Grunebaum, unpublished data) PNP, have all been investigated for the management of PNP deficiency. However, all these treatments are based on “cross-correction” of the toxic purine metabolites i.e., exit of nucleotides from the cytoplasm of cells in accordance to the purine concentration gradients. An alternative approach might be needed for the neurological deficits, such as delivery of PNP across the blood brain barrier and into the cells with the HIV-TAT protein transduction domain (Toro and Grunebaum 2006) or a modified diphtheria toxin (Auger et al. 2015). Finally, an exciting recent development in the use of iPSC is the creation of 3 dimensional brain organoids, which might provide a robust pre-clinical tool for assessing management strategies for the brain abnormalities prior to initiation of patients’ trials.
Conclusion
In conclusion, we report here the generation and use of neuronal cells derived from iPSC to model the neurological abnormalities associated with human PNP deficiency, thereby providing an important tool for better understanding and management of this condition.
Abbreviations
- HSC
- hematopoietic stem cells
- iPSC
- induced pluripotent stem cells
- PNP
- purine nucleoside phsphorylase
- PNP−/−
- PNP-deficient
Acknowledgements
This work was supported in part by the Donald and Audrey Campbell Chair for Immunology Research (EG). This work was part of the MT requirements for Master in Science thesis, Institute of Medical Sciences, School of Graduate Sciences, University of Toronto, Toronto, Ontario.
REFERENCES
Allende M.L., Cook E.K., Larman B.C., Nugent A., Brady J.M., Golebiowski D., Sena-Esteves M., Tifft C.J., and Proia R.L. 2018. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 59(3):550–563.
Andrade L.N., Nathanson J.L., Yeo G.W., Menck C.F., and Muotri A.R. 2012. Evidence for premature aging due to oxidative stress in iPSCs from Cockayne syndrome. Hum. Mol. Genet. 21(17):3825–3834.
Arpaia E., Benveniste P., Di Cristofano A., Gu Y., Dalal I., Kelly S., Hershfield M., Pandolfi P.P., Roifman C.M., and Cohen A. 2000. Mitochondrial basis for immune deficiency. Evidence from purine nucleoside phosphorylase-deficient mice. J. Exp. Med. 191(12):2197–2208.
Auger A., Park M., Nitschke F., Minassian L.M., Beilhartz G.L., Minassian B.A., and Melnyk R.A. 2015. Efficient delivery of structurally diverse protein cargo into mammalian cells by a bacterial toxin. Mol. Pharm. 12(8):2962–2971.
Baguette C., Vermylen C., Brichard B., Louis J., Dahan K., Vincent M.F., and Cornu G. 2002. Persistent developmental delay despite successful bone marrow transplantation for purine nucleoside phosphorylase deficiency. J. Pediatr. Hematol. Oncol. 24(1):69–71.
Barral S. and Kurian M.A. 2016. Utility of induced pluripotent stem cells for the study and treatment of genetic diseases: Focus on childhood neurological disorders. Front. Mol. Neurosci. 9:78.
Chailangkarn T., Trujillo C.A., Freitas B.C., Hrvoj-Mihic B., Herai R.H., Yu D.X., Brown T.T., Marchetto M.C., Bardy C., McHenry L., Stefanacci L., Järvinen A., Searcy Y.M., DeWitt M., Wong W., Lai P., Ard M.C., Hanson K.L., Romero S., Jacobs B., Dale A.M., Dai L., Korenberg J.R., Gage F.H., Bellugi U., Halgren E., Semendeferi K., and Muotri A.R. 2016. A human neurodevelopmental model for Williams syndrome. Nature. 536(7616):338–343.
Cheung A.Y., Horvath L.M., Grafodatskaya D., Pasceri P., Weksberg R., Hotta A., Carrel L., and Ellis J. 2011. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 20(11):2103–2115.
Cohen A., Grunebaum E., Arpaia E., and Roifman C.M. 2000. Immunodeficiency caused by purine nucleoside phosphorylase deficiency. Immunol. Allergy Clin. 20(1):143–159.
Delicou S., Kitra-Roussou V., Peristeri J., Goussetis E., Vessalas G., Rigatou E., Psychou F., Salavoura K., and Grafakos S. 2007. Successful HLA-identical hematopoietic stem cell transplantation in a patient with purine nucleoside phosphorylase deficiency. Pediatr. Transplant. 11(7):799–803.
Dror Y., Grunebaum E., Hitzler J., Narendran A., Ye C., Tellier R., Edwards V., Freedman M.H., and Roifman C.M. 2004. Purine nucleoside phosphorylase deficiency associated with a dysplastic marrow morphology. Pediatr. Res. 55(3):472–477.
Grunebaum E., Cohen A., and Roifman C.M. 2013. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies. Curr. Opin. Allergy Clin. Immunol. 13(6):630–638.
Grunebaum E., Zhang J., and Roifman C.M. 2004. Novel mutations and hot-spots in patients with purine nucleoside phosphorylase deficiency. Nucleosides Nucleotides Nucleic Acids. 23(8–9):1411–1415.
Jinnah H.A., Gage F.H., and Friedmann T. 1990. Animal models of Lesch-Nyhan syndrome. Brain Res. Bull. 25(3):467–475.
Liao P., Toro A., Min W., Lee S., Roifman C.M., and Grunebaum E. 2008. Lentivirus gene therapy for purine nucleoside phosphorylase deficiency. J. Gene Med. 10(12):1282–1293.
Mansouri A., Min W., Cole C.J., Josselyn S.A., Henderson J.T., van Eede M., Henkelman R.M., Ackerley C., Grunebaum E., and Roifman C.M. 2012. Cerebellar abnormalities in purine nucleoside phosphorylase deficient mice. Neurobiol. Dis. 47(2):201–209.
Mertens J., Marchetto M.C., Bardy C., and Gage F.H. 2016. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17(7):424–437.
Ozkinay F., Pehlivan S., Onay H., van den Berg P., Vardar F., Koturoglu G., Aksu G., Unal D., Tekgul H., Can S., and Ozkinay C. 2007. Purine nucleoside phosphorylase deficiency in a patient with spastic paraplegia and recurrent infections. J. Child Neurol. 22(6):741–743.
Papinazath T., Min W., Sujiththa S., Cohen A., Ackerley C., Roifman C.M., and Grunebaum E. 2011. Effects of purine nucleoside phosphorylase deficiency on thymocyte development. J. Allergy Clin. Immunol. 128(4):854–863.e1.
Patel M. and Yang S. 2010. Advances in reprogramming somatic cells to induced pluripotent stem cells. Stem Cell Rev. 6(3):367–380.
Rich K.C., Majias E., and Fox I.H. 1980. Purine nucleoside phosphorylase deficiency: Improved metabolic and immunologic function with erythrocyte transfusions. N. Engl. J. Med. 303(17):973–977.
Sauer A.V., Hernandez R.J., Fumagalli F., Bianchi V., Poliani P.L., Dallatomasina C., Riboni E., Politi L.S., Tabucchi A., Carlucci F., Casiraghi M., Carriglio N., Cominelli M., Forcellini C.A., Barzaghi F., Ferrua F., Minicucci F., Medaglini S., Leocani L., la Marca G., Notarangelo L.D., Azzari C., Comi G., Baldoli C., Canale S., Sessa M., D’Adamo P., and Aiuti A. 2017. Alterations in the brain adenosine metabolism cause behavioral and neurological impairment in ADA-deficient mice and patients. Sci. Rep. 7:40136.
Somech R., Lev A., Grisaru-Soen G., Shiran S.I., Simon A.J., and Grunebaum E. 2013. Purine nucleoside phosphorylase deficiency presenting as severe combined immune deficiency. Immunol. Res. 56(1):150–154.
Tabarki B., Yacoub M., Tlili K., Trabelsi A., Dogui M., and Essoussi A.S. 2003. Familial spastic paraplegia as the presenting manifestation in patients with purine nucleoside phosphorylase deficiency. J. Child Neurol. 18(2):140–141.
Toro A. and Grunebaum E. 2006. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J. Clin. Invest. 116(10):2717–2726.
Toro A., Paiva M., Ackerley C., and Grunebaum E. 2006. Intracellular delivery of purine nucleoside phosphorylase (PNP) fused to protein transduction domain corrects PNP deficiency in vitro. Cell. Immunol. 240(2):107–115.
Watson A.R., Evans D.I., Marsden H.B., Miller V., and Rogers P.A. 1981. Purine nucleoside phosphorylase deficiency associated with a fatal lymphoproliferative disorder. Arch. Dis. Child. 56(7):563–565.
Watson L.M., Wong M.M.K., Vowles J., Cowley S.A., and Becker E.B.E. 2018. A simplified method for generating Purkinje cells from human-induced pluripotent stem cells. Cerebellum.
Yeates L., Slatter M.A., and Gennery A.R. 2017. Infusion of sibling marrow in a patient with purine nucleoside phosphorylase deficiency leads to split mixed donor chimerism and normal immunity. Front. Pediatr. 5:143.
Yu Y., Arora A., Min W., Roifman C.M., and Grunebaum E. 2009. EdU incorporation is an alternative non-radioactive assay to [(3)H]thymidine uptake for in vitro measurement of mice T-cell proliferations. J. Immunol. Methods. 350(1–2):29–35.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 2 • June 2018
Pages: 49 - 56
History
Received: 20 February 2018
Accepted: 21 March 2018
Accepted manuscript online: 19 April 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MichaelTsui, JeremyBiro, JonathanChan, WeixianMin, and EyalGrunebaum. 2018. Use of induced pluripotent stem cells to investigate the effects of purine nucleoside phosphorylase deficiency on neuronal development. LymphoSign Journal.
5(2): 49-56. https://doi.org/10.14785/lymphosign-2018-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
