Chronic granulomatous disease due to different mutations in patients from the same consanguineous extended family
Abstract
Chronic granulomatous disease is a primary immunodeficiency disease caused by a genetic mutation in any of the 5 genes encoding the different components of the Nicotinamide Adenine Dinucleotide Phosphate reduced (NADPH)-Oxidase enzyme complex. Since primary immunodeficiency diseases are considered to be rare diseases, the genetic diagnosis of a certain primary immunodeficiency leads to the reasonable assumption that all patients with the same disease within the same family will have the same genetic mutation. We report 2 patients with chronic granulomatous disease from the same extended consanguineous family who had different genetic causes of their disease. Therefore, it is crucial to obtain a definitive genetic diagnosis of primary immunodeficiency disease even in patients from the same family, where the same genetic diagnosis is presumed to be the cause of the disease.
Statement of novelty: Genetic causes of chronic granulomatous disease may be different in patients from the same family.
Introduction
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency disease (PID) of impaired reactive oxygen species (ROS) production caused by inherited defects in the Nicotinamide Adenine Dinucleotide Phosphate reduced (NADPH)-Oxidase enzyme complex. The NADPH oxidase complex generates reactive oxygen species (ROS), which are required for intracellular pathogen killing through pH changes and potassium influx (Reeves et al. 2002). Furthermore, defects in NADPH activity and lack of ROS formation also impair extracellular pathogen killing by neutrophil extracellular traps (Romao et al. 2015). Ineffective ROS generation in patients with CGD leads to recurrent infections and hyper-inflammation. Mutations in any of the 5 components of the NADPH complex cause CGD. Mutations in the CYBB gene encoding the gp91phox protein causes X-linked CGD, whereas bi-allelic mutations in CYBA, NCF1, NCF2, and NCF4 genes encoding p22phox, p47phox, p67phox, and p40phox proteins, respectively, cause autosomal recessive (AR) CGD (reviewed in Leiding and Holland 2012).
In patients with typical clinical features compatible with CGD, the diagnosis of CGD can be reached by demonstrating lack of ROS production in neutrophils. However, establishing an unequivocal diagnosis of CGD requires genetic testing. Reaching a genetic diagnosis is clinically important since most X-linked CGD patients have a more severe phenotype and a worse prognosis compared with p47phox-deficient CGD; therefore, offering a curative HSCT is based not only on the clinical phenotype of the patient but also on the genetic cause of CGD (Leiding and Holland 2012). The somewhat milder phenotype in AR CGD compared to X-linked CGD, as manifested by increased survival, is associated with residual NADPH activity mostly in P47phox-deficient AR CGD (Kuhns et al. 2010).
CGD is a rare disease with a prevalence ranging from as high as 1:79 000 (Leiva et al. 2007) to 1:450 000 (Ǻhlin et al. 1995), but the prevalence and incidence are higher in the consanguineous populations in Israel. The latest report shows a birth incidence of 1.05 per 100 000 live births in the Jewish population versus 1.45 per 100 000 live births in the more consanguineous Arab Israeli population (Wolach et al. 2017). Therefore, the assumption that the diagnosis of 2 children with CGD in 1 consanguineous, extended family is caused by the same genetic mutation is reasonable. Nevertheless, we report on the diagnosis of 2 children, from the same extended family, with typical CGD, who eventually were diagnosed as having different genetic causes of CGD.
The genetic studies reported here were performed following an informed consent.
Case presentations
Patient 1
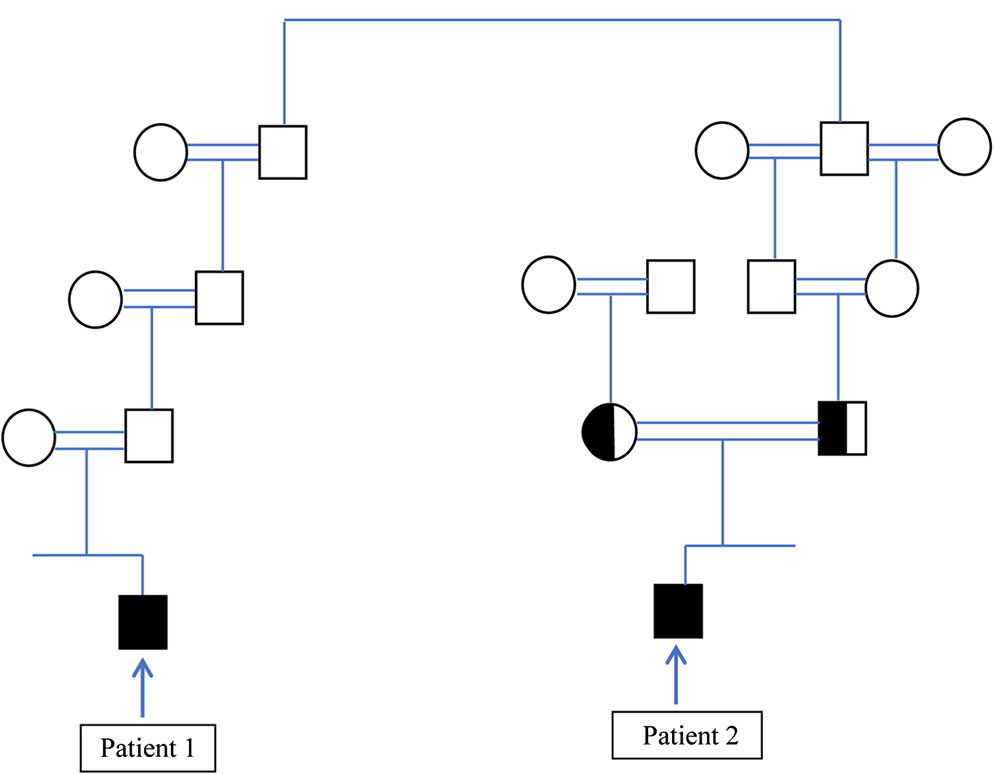
This male patient is currently 15 years old and born from a consanguineous marriage (Figure 1). He was healthy until 18 months of age when he presented with fever, ascites, and hepatosplenomegaly. He was treated with broad spectrum antibiotics and recovered. At the age of 5 years he was treated for severe anemia (hemoglobin 7 g/dL) with supplemental oral and intravenous iron, with only a partial response. At 6 years of age he was hospitalized due to cervical lymphadenitis, with a complete recovery after treatment with parenteral antibiotics. At 7 years of age he was again hospitalized for pneumonia with elevated inflammatory markers. A ground-glass appearance was evident on chest computed tomography; however, a lung biopsy was inconclusive. At 10 years of age he was again admitted due to cervical lymphadenopathy. He was treated with parenteral antibiotics and had only a partial improvement. At this time an immunological workup revealed no superoxide production in the dihydro-rhodamine (DHR) assay (stimulation index of 1.65 with only 1.27% activity compared to control), and the diagnosis of CGD was reached. At approximately the same time, patient 2 was also found to have CGD.
Figure 1:
Patient 2
This male patient is currently 14 years old. He was also born from a consanguineous marriage, and related to patient 1 (Figure 1). He did not receive any special medical attention until 8 years of age. He was then noted to have perianal aphthae and recurrent aphthous stomatitis of 2 years’ duration. Immunological workup revealed only residual NADPH activity with a stimulation index of 4.26 with only 3.2% activity compared to control, by the DHR assay, and the diagnosis of CGD was reached.
In these patients NADPH subunit analysis by immunoblotting revealed lack of gp91phox in patient 1 and lack of p47phox in patient 2. Genetic analysis revealed a de-novo mutation (the mother was not a carrier of the mutation) in the CYBB gene, c.742dupA, leading to a frameshift mutation and premature termination of protein synthesis (p. Ile248AsnfsTer36) in patient 1. A common homozygous delta-GT mutation in the NCF1 gene (c.75_76delGT) was discovered in patient 2, predicting a frameshift and premature termination of protein synthesis (p. Tyr26HisfsTer26). Following this diagnosis, 2 additional brothers of patient 2 were diagnosed as having the same mutation despite having a relatively benign clinical history with only minor, non-invasive infections.
Discussion
In this report we show that CGD can occur in the same extended family due to different genetic causes. It seems reasonable to assume that patients from the same family with a distinct PID phenotype will have the same genetic cause of disease, and if 1 patient in the family is diagnosed with a disease-causing mutation, then other patients from the same family will have the same mutation, particularly in highly consanguineous families. In this report we show that this assumption may be erroneous. In many developing countries genetic testing for rare genetic diseases is not readily available, and the costs of genetic testing may be charged to the family and not performed for this reason. In such instances, genetic testing may be performed only for 1 patient and not for other family members, even though they may have the same (as yet undetected) disease. Indeed, after the diagnosis of patient 1, 2 additional brothers were diagnosed as having CGD even though they did not have typical infections. If we would have assumed that the mutation discovered in patient 2 is the same disease-causing mutation in patient 1, we would have missed the correct genetic diagnosis.
Although different rare genetic primary immunodeficiency diseases may co-exist in the same family (Broides et al. 2009) and sometimes even in the same patient (Ehlayel et al. 2008), the present report adds to our knowledge by showing that the same primary immunodeficiency, in this case CGD, can be due to different genetic causes even within 1 extended family.
It is noteworthy that patient 2 had some residual NADPH activity compared to patient 1 who had virtually no NADPH activity, and indeed patient 2 displayed a milder phenotype compared with patient 1. These different clinical courses can be expected based on the residual NADPH activity in patient 2 (Kuhns et al. 2010).
Identifying the genetic defect is essential and crucial. It will aid in offering prenatal diagnosis, early diagnosis and treatment to affected children and may have therapeutic implications. Such is the case in CGD, since patients with X-linked CGD who have a severe phenotype, and no residual NADPH activity may be offered a HSCT more readily than those with AR CGD who have a milder phenotype and some residual NADPH activity. Indeed, the clinical course of patient 1 with X-linked CGD was more severe than patient 2 with AR CGD.
This report shows the importance of reaching the correct genetic diagnosis in PID patients, even when performing such studies may seem redundant due to the assumption that patients with a similar phenotype from an extended consanguineous family should have the same genetic cause of their disease.
REFERENCES
Ǻhlin A., De Boer M., Roos D., Leusen J., Smith C.I., Sundin U., Rabbani H., Palmblad J., and Elinder G. 1995. Prevalence, genetics and clinical presentation of chronic granulomatous disease in Sweden. Acta Paediatr. 84:1386–1394.
Broides A., Shubinsky G., Parvari R., Grimbacher B., Somech R., Garty B.Z., and Levy J. 2009. MHC class 2 deficiency and X-linked agammaglobulinaemia in a consanguineous extended family. Int. J. Immunogenet. 36(4):223–226.
Ehlayel M., de Beaucoudrey L., Fike F., Nahas S.A., Feinberg J., Casanova J.L., and Gatti R.A. 2008. Simultaneous presentation of 2 rare hereditary immunodeficiencies: IL-12 receptor beta1 deficiency and ataxia-telangiectasia. J. Allergy Clin. Immunol. 122(6):1217–1219.
Kuhns D.B., Alvord W.G., Heller T., Feld J.J., Pike K.M., Marciano B.E., Uzel G., DeRavin S.S., Priel D.A., Soule B.P., Zarember K.A., Malech H.L., Holland S.M., and Gallin J.I. 2010. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl. J. Med. 363:2600–2610.
Leiding, J.W., Holland, S.M. 2012. Chronic granulomatous disease. In: GeneReviews®. Edited by M.P. Adam, H.H. Ardinger, R.A. Pagon, S.E. Wallace, L.J.H. Bean, K. Stephens, and A. Amemiya. Seattle, WA: University of Washington; 1993–2018.
Leiva L.E., Zelazco M., Oleastro M., Carneiro-Sampaio M., Condino-Neto A., Costa-Carvalho B.T., Grumach A.S., Quezada A., Patiño P., Franco J.L., Porras O., Rodríguez F.J., Espinosa-Rosales F.J., Espinosa-Padilla S.E., Almillategui D., Martínez C., Tafur J.R., Valentín M., Benarroch L., Barroso R., Sorensen R.U., and Latin American Group for Primary Immunodeficiency Diseases. 2007. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J. Clin. Immunol. 27:101–108.
Reeves E.P., Lu H., Jacobs H.L., Messina C.G., Bolsover S., Gabella G., Potma E.O., Warley A., Roes J., and Segal A.W. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 416:291–297.
Romao S., Puente E.T., Nytko K.J., Siler U., Münz C., and Reichenbach J. 2015. Defective nuclear entry of hydrolases prevents neutrophil extracellular trap formation in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 136(6):1703–1706.e5.
Wolach B., Gavrieli R., de Boer M., van Leeuwen K., Berger-Achituv S., Stauber T., Ben Ari J., Rottem M., Schlesinger Y., Grisaru-Soen G., Abuzaitoun O., Marcus N., Zion Garty B., Broides A., Levy J., Stepansky P., Etzioni A., Somech R., and Roos D. 2017. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am. J. Hematol. 92:28–36.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 2 • June 2018
Pages: 57 - 60
History
Received: 22 February 2018
Accepted: 3 April 2018
Accepted manuscript online: 15 April 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
ArnonBroides, RonitGavrieli, JacovLevy, RachelLevy, NuritHadad, DirkRoos, BaruchWolach, and AmitNahum. 2018. Chronic granulomatous disease due to different mutations in patients from the same consanguineous extended family. LymphoSign Journal.
5(2): 57-60. https://doi.org/10.14785/lymphosign-2018-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
