Monocytosis in a patient with a novel GATA2 mutation
Abstract
GATA2-associated disorders include: (i) monocytopenia with mycobacterial infections (MonoMAC); (ii) dendritic cell, monocyte, B and NK lymphoid deficiency; (iii) familial myelodysplastic syndrome (MDS) and acute myeloid leukemia; and (iv) congenital deafness with lower limb lymphedema deficiency (Emberger syndrome). Markedly reduced or absent monocytes have been considered as the hallmark of the disease. Here we report on a patient that presented in infancy with hearing loss and lymphedema. By 4 years of age the patient developed acne, disseminated warts, lymphadenopathy, and MDS, yet with increased monocyte as well as normal NK- and B-cell numbers. The patient was found to have a novel mutation in GATA2 that was predicted to disrupt the C-terminal zinc finger. Importantly, and in contrast to common concepts, GATA2-associated syndromes might present with monocytosis.
Statement of novelty: We describe a novel mutation in GATA2 associated with monocytosis.
Introduction
The GATA2 gene, located at chromosome 3q21.3, encodes a dual zinc-finger transcription factor, known as GATA binding protein 2 that binds to the promoter and regulatory regions of target genes in the nucleus. GATA2 regulates transcription of genes involved in the development and proliferation of diverse cell lineages, including hematopoietic, endothelial, placental, neuronal, etc. (Bresnick et al. 2010). The impaired function of hematopoietic stem cells from GATA2 +/− mice (Rodrigues et al. 2005) and the association of hematological malignancies with translocation of GATA2 enhancer elements (Gröschel et al. 2014) suggest a dose-dependent effect for GATA2 on hematopoiesis. Indeed, heterozygous GATA2 mutations were recently identified among patients with diverse hematological and nonhematological manifestations. These conditions have been characterized as: (i) monocytopenia and mycobacterial infections (MonoMAC) syndrome (Hsu et al. 2011); (ii) dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency (Dickinson et al. 2011); (iii) familial myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) (Hahn et al. 2011); and (iv) primary lymphedema, congenital deafness, and MDS (Emberger syndrome) (Ostergaard et al. 2011). Additionally, GATA2 mutations predispose for pulmonary alveolar proteinosis, disseminated warts and human papillomavirus-associated squamous cell cancer (Hsu et al. 2011), childhood neutropenia (Pasquet et al. 2013), as well as NK-cell deficiency (Mace et al. 2013). Recent analysis of 57 patients with GATA2 mutations found a high correlation (p < 0.001) between monocytopenia, B, NK, and CD4 lymphocytopenia and the presence of disease (Spinner et al. 2014), emphasizing the importance of measuring monocyte numbers for the diagnosis of this condition. Indeed, 78.9% of this cohort had reduced monocyte numbers, 15.7% had normal monocyte numbers, and only 5.3% of the patients had increased monocyte numbers (Spinner et al. 2014). However, this cohort as well as other reports of GATA2-associated disorders included mostly teenagers and adult patients (Pasquet et al. 2013; Dickinson et al. 2014).
Here we report on a patient that presented in infancy with severe hearing loss and lymphedema. By 4 years of age the patient developed acne, disseminated warts, lymphadenopathy, and myelodysplastic syndrome. This patient had long-standing monocytosis with a novel GATA2 mutation.
Case report
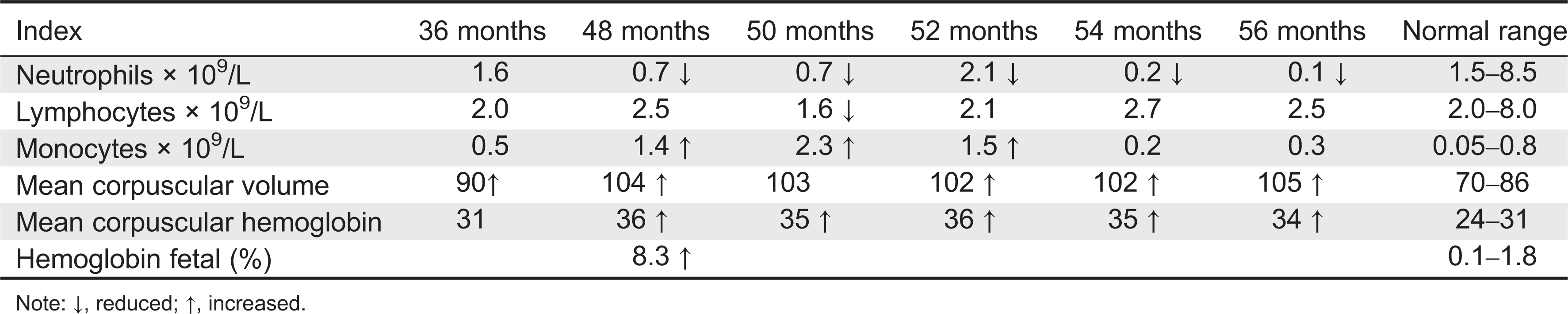
The Research Ethics Board at the Hospital for Sick Children, Toronto, Ontario, approved the study, and a written informed consent for the research was obtained from the parents. The patient was the second daughter born to nonconsanguineous parents after a full-term pregnancy that was complicated by maternal heart arrhythmia. Immediately after birth, apnea episodes were noted, and the patient was treated for 7 days for presumed pneumonia and sepsis with a gentamicin-containing regimen. At 2 months of age the patient was diagnosed with profound sensory neural hearing deficit, for which she received cochlear implants. Analysis of several genes associated with hearing loss, including GJB2, GJB6, SLC26A4, MTRNR1, and MTTS1 was normal. The patient also had pulmonary artery narrowing that resolved. At 3 months of age lymphoedema was noted, which initially involved the left foot and right hand and progressed to all extremities by 6 months of age. A lymphogram demonstrated an absent migration of lymph in the upper left extremity as well as slow migration in the right upper and left lower extremities. A micro-array demonstrated a 458 kb insertional duplication of chromosome 9Q34.3 of unknown significance. At 4 years of age, because of unusual facial acne (Figure 1A), multiple planter warts (Figure 1B), recurrent respiratory and ear infections, as well as persistent lymphadenopathy the patient was referred for immune evaluation. Laboratory findings included normal immunoglobulin levels, production of antibodies, T-, B-, and NK-cell numbers as well as normal T-cell response to stimulation (Table 1). At that time, the patient was also noted to have intermittent neutropenia, markedly increased mean corpuscular volume and mean corpuscular hemoglobin, and elevated hemoglobin F, whereas monocytes were persistently increased or normal (Table 2). Bone marrow examinations revealed mildly hypocellular particles with adequate trilineage hematopoiesis and no increase in blasts. Cytogenetic analysis by G banding and interphase fluorescent in situ hybridization showed monosomy 7 in the majority of the cells. A diagnosis of MDS was made. Because of the patient's constellation of symptoms, the GATA2 gene was interrogated in the patients’ peripheral blood mononuclear cells. Sequencing demonstrated a heterozygous novel c.1041delC mutation (Figure 2) immediately upstream to the C-terminal zinc-finger domain. The mutation is predicted to cause a reading frame shift and to later stop codon 39 amino acids (Figure 3). The mutation was not present in the patient's parents or sister. Although the mutation was not demonstrated to be a germ line, the rarity of such somatic mutations in MDS and the constitutional features of the patient suggest that the GATA2 mutation was the cause of the disease.
Figure 1:
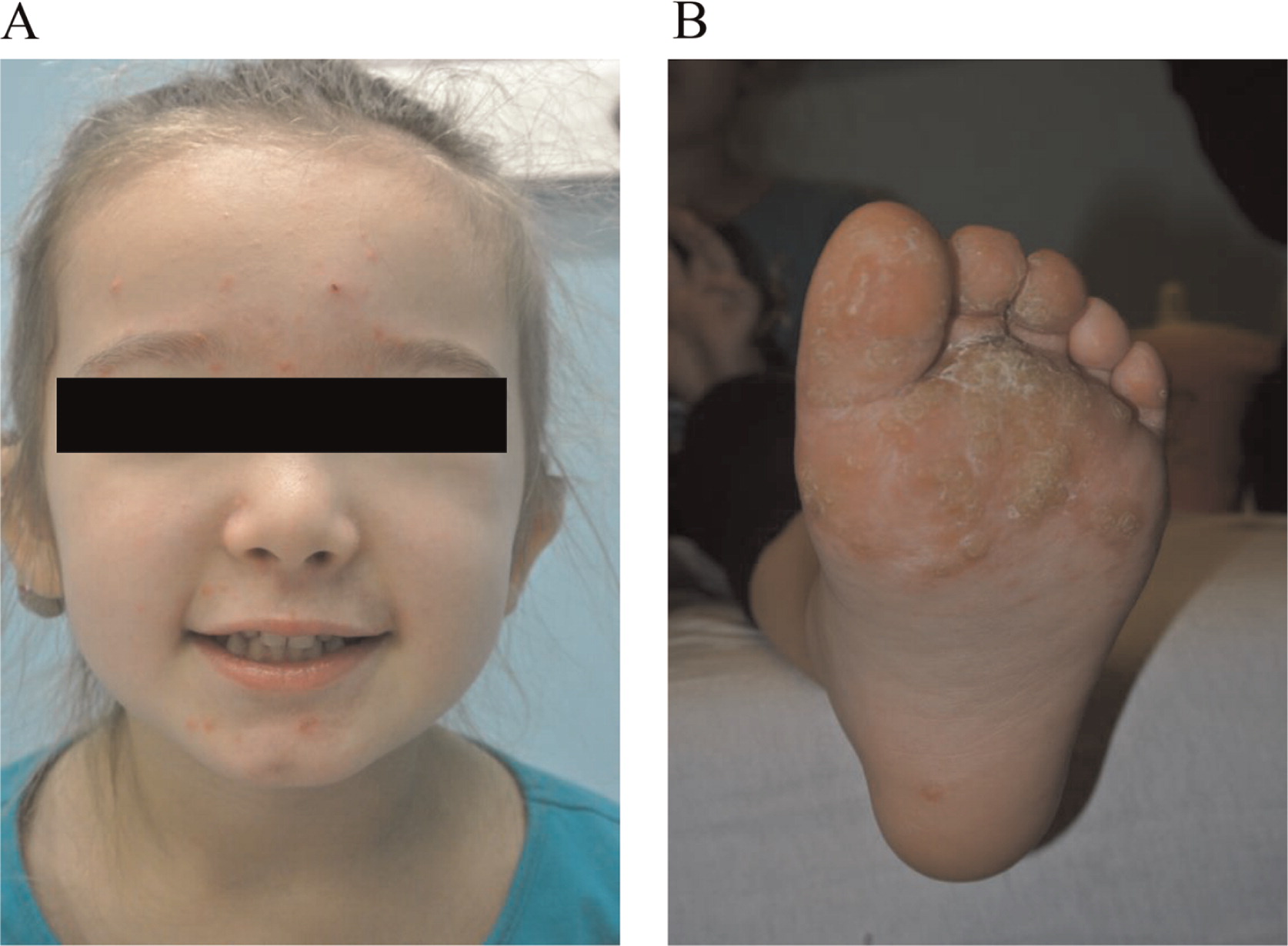
Figure 2:
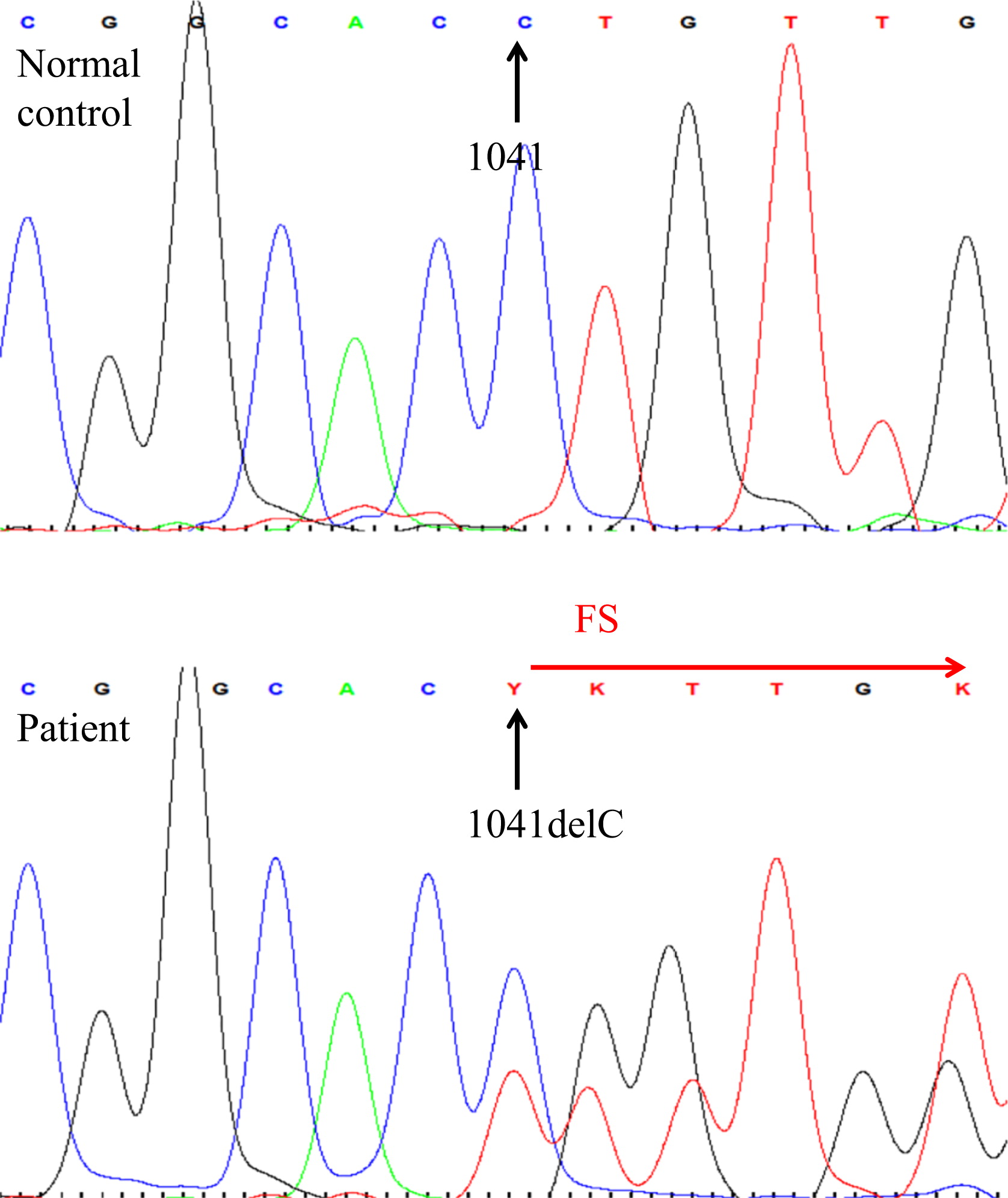
Figure 3:
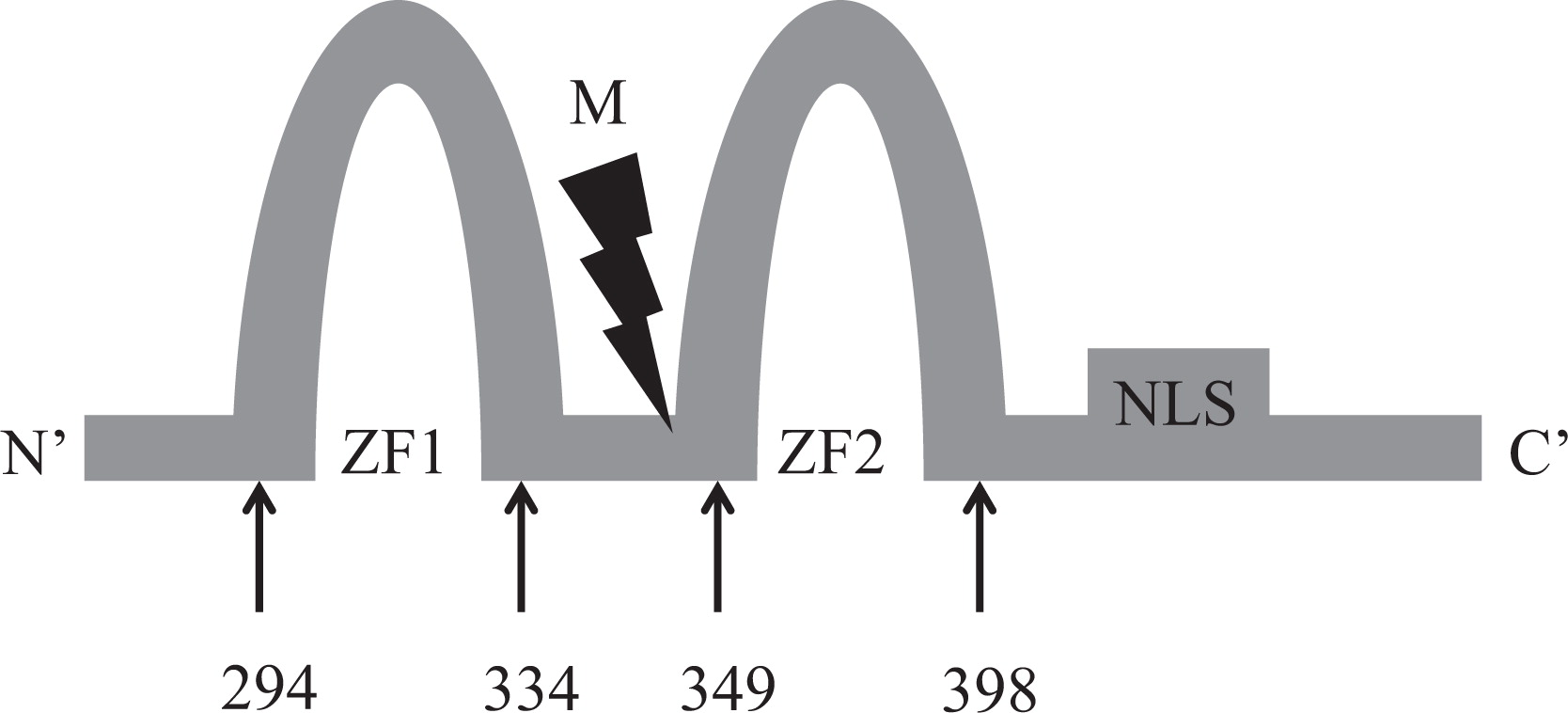
Table 1:
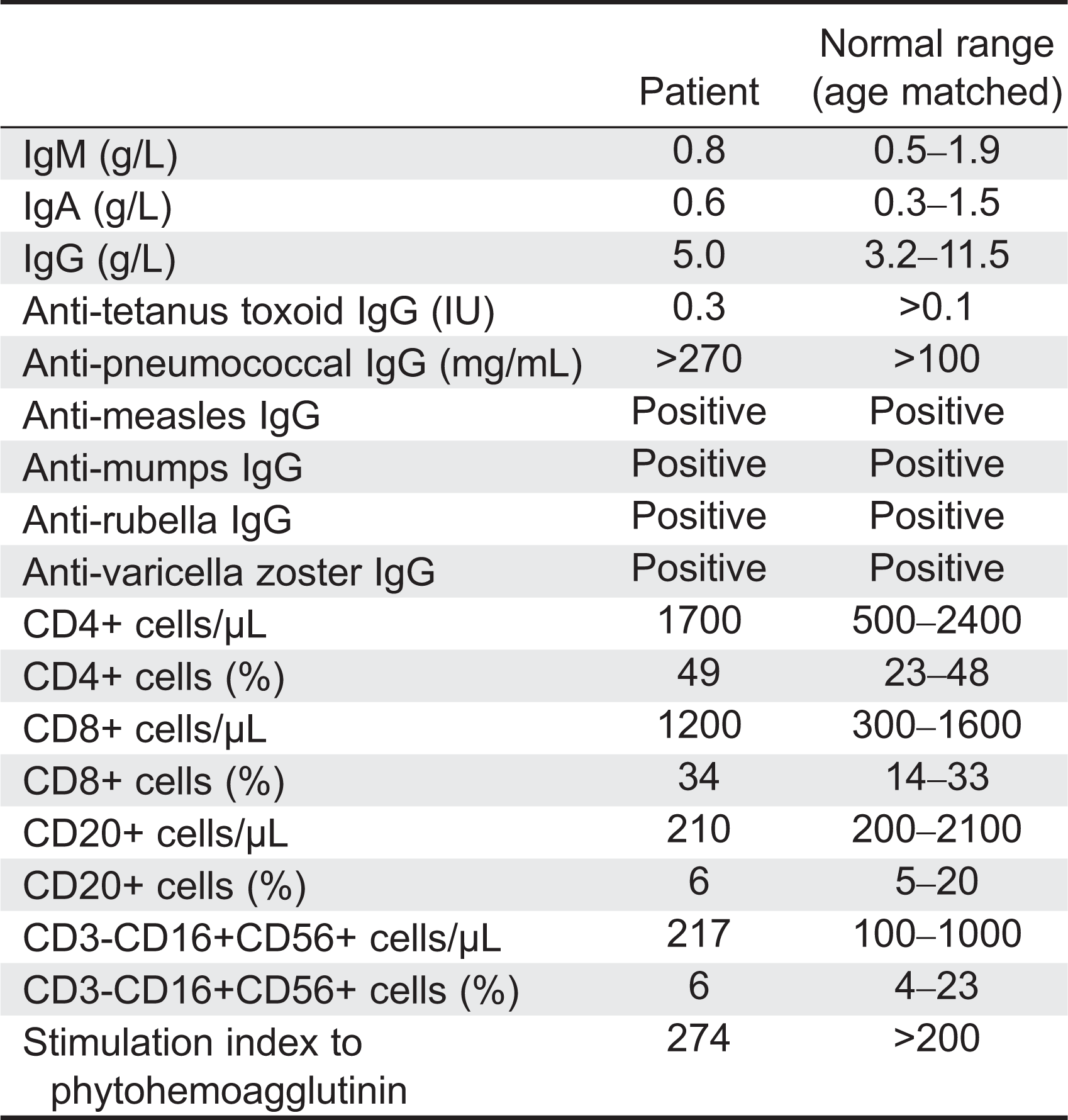
Table 2:
Because of the MDS with progressive neutropenia and ominous cytogenetics, the patient underwent bone marrow transplantation using a 10/10 human leukocyte antigen (HLA) matched unrelated donor at 56 months of age. The myeloablative conditioning regimen comprised fludarabine (160 mg/m2) and busulfan (3.2 mg·kg−1·dose−1, adjusted to achieve area under the curve of 3600–6000 micromol/litre/minute × 4 doses).
Discussion
Many patients with GATA2 mutations have reduced lymphocytes and severe monocytopenia. Therefore, low monocyte numbers have been traditionally considered the sine qua non for the diagnosis of GATA2 deficiency. Appropriate GATA2 expression is critical for the maturation of hematopoietic cells (Bresnick et al. 2010); hence, the common hypothesis is that developmental abnormality results in reduced cell numbers. However, the patient described here had elevated monocyte counts as well as the normal NK- and B-cell numbers. It is possible that the monocytosis is related to the development of the myelodysplasia, as already suggested in a recent cross-sectional analysis of 24 patients' with GATA2 deficiency (Dickinson et al. 2014), although monocytopenia were common and profound among patients with GATA2 defects even in the setting of MDS (Spinner et al. 2014). Also, the normal monocyte numbers observed in our patient at 36 months of age, prior to establishing the diagnosis of MDS, might have been a harbinger of the bone marrow abnormalities, as reflected by the increased mean corpuscular volume at that age. Regardless of the etiology of monocytosis, our patient illustrates the importance of investigating GATA2 even in the absence of monocytopenia, and considering myelodysplasia among patients with GATA2 defects who have normal or increased monocyte numbers.
The original publication by Emberger et al. (1979), as well as subsequent studies of patients with Emberger syndrome and GATA2 deficiency, described lymphedema that often appeared in childhood and adolescence (Mansour et al. 2010). In contrast, lymphedema was already detected in our patient at 3 months of age and she suffered from disseminated warts and significant hematopoietic abnormalities by 4 years of age. Although a clear genotype–phenotype correlation has not been established in GATA2 deficiency, a recent report suggested that mutations causing frame shift and disruption of the C-terminal zinc finger, such as the novel one identified in our patient, are associated with a younger age of presentation and higher clinical score (Dickinson et al. 2014). Future studies involving a larger number of patients will help clarify whether patients with frame-shift mutations and haplo-insufficiency of the C-terminal zinc finger should be managed differently than patients with amino acid substitutions.
Optimal treatment for GATA2 deficiency has not been established. Our patient and many other GATA2-deficient patients suffered from repeated infections and disseminated warts. Yet, T-cell numbers and functions as well as production of antibodies are often preserved. Hence, antibiotic and (or) immunoglobulin prophylaxis are unlikely to be of significant clinical benefit. It is possible that monocyte function might be improved by use of a granulocyte–macrophage colony stimulating factor, similar to the benefit observed among patients with other macrophage defects (Wang et al. 2005; Greenhill and Kotton 2009). However, this strategy has not been reported in GATA2-deficient patients and might be associated with significant risk because of the potential to accelerate propensity for MDS or AML in genetically altered stem cells, as suggested for other growth factors and patients with marrow abnormalities (Beekman and Touw 2010). Hematopoietic stem cell transplantations (HSCT) with myeloablative or nonmyeloablative conditioning have been utilized in GATA2-deficient patients, primarily because of hematological malignancies and progressive immunodeficiency (Cuellar-Rodriguez et al. 2011; Pasquet et al. 2013; Grossman et al. 2014). Our patient received HSCT at a very young age, before the development of malignancy and severe infections, albeit after the appearance of diffuse lymphedema, a severe hearing defect, and MDS with monosomy 7. Accordingly, long-term follow up of our patient will provide a unique opportunity to assess whether HSCT can prevent the complications associated with GATA2 deficiency including pulmonary alveolar proteinosis and gynecological malignancies.
In conclusion, GATA2 defects should be investigated in patients suffering from early hearing loss, lymphedema, recurrent infections, and (or) hematological abnormalities, particularly MDS or familial AML, even if monocytes and lymphocyte subpopulations are not affected. The best treatment for GATA2-deficient patients needs to be determined.
Acknowledgments
The authors thank Dr. C.M. Roifman for performing the molecular analysis of the GATA2 gene. E.G. is supported by the Donald and Audrey Chair for Immunology Research, Hospital for Sick Children and the University of Toronto.
References
Beekman R. and Touw I.P. G-CSF and its receptor in myeloid malignancy Blood. 2010 115 25 5131 -5136
Bresnick E.H., Lee H.Y., Fujiwara T., Johnson K.D., and Keles S. GATA switches as developmental drivers J. Biol. Chem. 2010 285 41 31087 -31093
Cuellar-Rodriguez J., Gea-Banacloche J., Freeman A.F., Hsu A.P., Zerbe C.S., Calvo K.R., Wilder J., Kurlander R., Olivier K.N., Holland S.M., and Hickstein D.D. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency Blood. 2011 118 13 3715 -3720
Dickinson R.E., Griffin H., Bigley V., Reynard L.N., Hussain R., Haniffa M., Lakey J.H., Rahman T., Wang X.N., McGovern N., Pagan S., Cookson S., McDonald D., Chua I., Wallis J., Cant A., Wright M., Keavney B., Chinnery P.F., Loughlin J., Hambleton S., Santibanez-Koref M., and Collin M. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency Blood. 2011 118 10 2656 -2658
Dickinson R.E., Milne P., Jardine L., Zandi S., Swierczek S.I., McGovern N., Cookson S., Ferozepurwalla Z., Langridge A., Pagan S., Gennery A., Heiskanen-Kosma T., Hämäläinen S., Seppänen M., Helbert M., Tholouli E., Gambineri E., Reykdal S., Gottfreðsson M., Thaventhiran J.E., Morris E., Hirschfield G., Richter A.G., Jolles S., Bacon C.M., Hambleton S., Haniffa M., Bryceson Y., Allen C., Prchal J.T., Dick J.E., Bigley V., and Collin M. The evolution of cellular deficiency in GATA2 mutation Blood. 2014 123 6 863 -874
Emberger J.M., Navarro M., Dejean M., and Izarn P. Deaf-mutism, lymphedema of the lower limbs and hematological abnormalities (acute leukemia, cytopenia) with autosomal dominant transmission J. Genet. Hum. 1979 27 3 237 -245
Greenhill S.R. and Kotton D.N. Pulmonary alveolar proteinosis: a bench-to-bedside story of granulocyte-macrophage colony-stimulating factor dysfunction Chest. 2009 136 2 571 -577
Grossman J., Cuellar-Rodriguez J., Gea-Banacloche J., Zerbe C., Calvo K., Hughes T., Hakim F., Cole K., Parta M., Freeman A., Holland S.M., and Hickstein D.D. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014
Gröschel S., Sanders M.A., Hoogenboezem R., de Wit E., Bouwman B.A., Erpelinck C., van der Velden V.H., Havermans M., Avellino R., van Lom K., Rombouts E.J., van Duin M., Döhner K., Beverloo H.B., Bradner J.E., Döhner H., Löwenberg B., Valk P.J., Bindels E.M., de Laat W., and Delwel R. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia Cell. 2014 157 2 369 -381
Hahn C.N., Chong C.E., Carmichael C.L., Wilkins E.J., Brautigan P.J., Li X.C., Babic M., Lin M., Carmagnac A., Lee Y.K., Kok C.H., Gagliardi L., Friend K.L., Ekert P.G., Butcher C.M., Brown A.L., Lewis I.D., To L.B., Timms A.E., Storek J., Moore S., Altree M., Escher R., Bardy P.G., Suthers G.K., D'Andrea R.J., Horwitz M.S., and Scott H.S. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia Nat. Genet. 2011 43 10 1012 -1017
Hsu A.P., Sampaio E.P., Khan J., Calvo K.R., Lemieux J.E., Patel S.Y., Frucht D.M., Vinh D.C., Auth R.D., Freeman A.F., Olivier K.N., Uzel G., Zerbe C.S., Spalding C., Pittaluga S., Raffeld M., Kuhns D.B., Ding L., Paulson M.L., Marciano B.E., Gea-Banacloche J.C., Orange J.S., Cuellar-Rodriguez J., Hickstein D.D., and Holland S.M. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome Blood. 2011 118 10 2653 -2655
Mace E.M., Hsu A.P., Monaco-Shawver L., Makedonas G., Rosen J.B., Dropulic L., Cohen J.I., Frenkel E.P., Bagwell J.C., Sullivan J.L., Biron C.A., Spalding C., Zerbe C.S., Uzel G., Holland S.M., and Orange J.S. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset Blood. 2013 121 14 2669 -2677
Mansour S., Connell F., Steward C., Ostergaard P., Brice G., Smithson S., Lunt P., Jeffery S., Dokal I., Vulliamy T., Gibson B., Hodgson S., Cottrell S., Kiely L., Tinworth L., Kalidas K., Mufti G., Cornish J., Keenan R., Mortimer P., Murday V., and Lymphoedema Research Consortium Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases Am. J. Med. Genet. A. 2010 152A 9 2287 -2296
Ostergaard P., Simpson M.A., Connell F.C., Steward C.G., Brice G., Woollard W.J., Dafou D., Kilo T., Smithson S., Lunt P., Murday V.A., Hodgson S., Keenan R., Pilz D.T., Martinez-Corral I., Makinen T., Mortimer P.S., Jeffery S., Trembath R.C., and Mansour S. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat. Genet. 2011 43 10 929 -931
Pasquet M., Bellanné-Chantelot C., Tavitian S., Prade N., Beaupain B., Larochelle O., Petit A., Rohrlich P., Ferrand C., Van Den Neste E., Poirel H.A., Lamy T., Ouachée-Chardin M., Mansat-De Mas V., Corre J., Récher C., Plat G., Bachelerie F., Donadieu J., and Delabesse E. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia Blood. 2013 121 5 822 -829
Rodrigues N.P., Janzen V., Forkert R., Dombkowski D.M., Boyd A.S., Orkin S.H., Enver T., Vyas P., and Scadden D.T. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis Blood. 2005 106 2 477 -484
Spinner M.A., Sanchez L.A., Hsu A.P., Shaw P.A., Zerbe C.S., Calvo K.R., Arthur D.C., Gu W., Gould C.M., Brewer C.C., Cowen E.W., Freeman A.F., Olivier K.N., Uzel G., Zelazny A.M., Daub J.R., Spalding C.D., Claypool R.J., Giri N.K., Alter B.P., Mace E.M., Orange J.S., Cuellar-Rodriguez J., Hickstein D.D., and Holland S.M. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity Blood. 2014 123 6 809 -821
Wang J., Mayer L., and Cunningham-Rundles C. Use of GM-CSF in the treatment of colitis associated with chronic granulomatous disease J. Allergy Clin. Immunol. 2005 115 5 1092 -1094
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 2 • June 2015
Pages: 85 - 90
History
Received: 9 October 2014
Accepted: 11 December 2014
Accepted manuscript online: 11 December 2014
Version of record online: 11 December 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Michaela Cada, Irene Lara-Corrales, Yigal Dror, Stephen Feanny, Vy Hong-Diep Kim, and Eyal Grunebaum. 2015. Monocytosis in a patient with a novel GATA2 mutation. LymphoSign Journal.
2(2): 85-90. https://doi.org/10.14785/lpsn-2014-0022
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
