A prospective outcome study of patients with profound combined immunodeficiency (P-CID)
Abstract
This is a prospective outcome study of patients with profound combined immunodeficiency (P-CID) (study number DRKS00000497).
Combined immunodeficiencies (CID) are a heterogeneous group of inherited immune disorders with impaired T-cell development and (or) function manifesting through increased susceptibility to infections and (or) immune dysregulation. They can be delineated from severe CID (SCID) by their manifestation beyond the first year of life. Profound CID (P-CID) is a potentially life-threatening form of CID, in which stem cell transplant (SCT) is a relevant consideration at diagnosis.
The primary objective of the study is to provide natural history data on patients with P-CID, irrespective of whether they undergo hematopoietic stem cell transplant (HSCT) or not. The goals are to determine survival, the frequency of severe events, and quality of life (QOL) 5 years after study inclusion.
The secondary objective is to develop a risk model for P-CID patients. The model is developed from a set of clinical and laboratory parameters obtained at diagnosis, at study inclusion, and yearly thereafter.
The tertiary objectives of this study are to determine the effects of donor, recipient, and treatment factors on the outcome of HSCT. The goal is to determine the quality of engraftment and immunological reconstitution and to determine the effects of these parameters on clinical outcome.
The main hypothesis is that P-CID patients undergoing early HSCT have a better 5-year survival rate than patients who undergo late HSCT or are not transplanted.
This is a prospective multi-centre international cohort study (observational study).
Enrolled patients will be evaluated and treated according to local institutional protocols. They will receive comparable baseline and follow-up evaluations across all participating centres, irrespective of the therapeutic strategy at the individual site.
There will be at least 6 study visits (scheduled yearly) for all patients. Because of the variable history prior to study inclusion, a morbidity score is determined for each patient at study visit 1. For those patients undergoing HSCT, an additional 6 month post-HSCT visit will be scheduled. The study visits will document immunological parameters, severe events including major infections, and major manifestations of immune dysregulation, severe transplant-related events, and QOL.
Introduction
Background and scientific basis
Definition and natural history of combined immunodeficiencies (2.1.11)
Although enormous progress has been made in the genetic definition of primary immunodeficiency diseases (Pessach et al. 2009), many patients still remain without molecular diagnosis. This renders prognosis and treatment decisions difficult. Combined immunodeficiencies (CID) are defined by their impaired T- and B-cell immunity leading to increased susceptibility to bacterial, viral, and opportunistic infections, disorders of immune regulation, and malignancies (Buckley 2000a; International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies et al. 2009; Notarangelo 2010). They include molecularly characterized immunodeficiency syndromes such as bare lymphocyte syndrome, WAS, CD40L deficiency, XLP, or Dock-8 deficiency (Allen et al. 1993; Derry et al. 1994; Coffey et al. 1998; Villard et al. 2001; Zhang et al. 2009), where observations on outcome and prognosis in a molecularly defined patient group provide a helpful framework for treatment decisions. However, CIDs also include a heterogeneous group of molecularly undefined immunodeficiencies, where observations on outcomes are limited and the decisions about therapy are much more difficult. A significant proportion of these patients are at risk for life-threatening infections and severe immune dysregulation (profound-CID, P-CID), and stem cell transplantation (SCT) is therefore a relevant therapeutic consideration. However, since there are no systematic outcome studies in P-CID patients, the criteria for SCT and its appropriate time point are poorly defined.
CIDs can be diagnosed in early childhood, in adolescence, or even in adulthood. When manifesting in childhood, CID must be differentiated from severe combined immunodeficiency (SCID), which by its clinical definition always presents in the first year of life. There is little debate that SCT represents the treatment of choice for this life-threatening disorder. There is more uncertainty about the treatment of patients with hypomorphic mutations or a genetic reversion in a SCID-causing gene, which can present with a much milder immunodeficiency. These variants are often associated with features of immune dysregulation in addition to infection susceptibility and have been termed “atypical” SCID (Notarangelo et al. 2000; Liston et al. 2008; Niehues et al. 2010). The exclusion of atypical SCID in a CID patient is not easy and frequently requires extensive genetic work-up. From a clinical viewpoint, the distinction is of limited help. Although the diagnosis of a molecular defect is frequently used as an argument in favour of SCT, this is not based on evidence. Currently, the prognosis for atypical SCID is as unclear as for profound CID of unknown molecular cause.
In particular when manifesting later in life, CID must be differentiated from common variable immunodeficiency (CVID). It is well recognized that a relevant proportion of patients with a clinical diagnosis of CVID (up to 10%) also presents with low numbers of T cells and (or) impaired T-cell function leading to opportunistic infections and many of those develop symptoms of immune dysregulation (Malphettes et al. 2009). The term “late-onset combined immunodeficiency” (LOCID) has recently been introduced to describe these patients (Malphettes et al. 2009). Again, exclusion of known molecular causes of immunodeficiency is difficult in these patients, owing to significant clinical and immunological overlap, in particular in the case of hypomorphic mutations. Thus, some patients with LOCID have been diagnosed as atypical SCID. Because they show an increased mortality, there is doubt that the usual treatment concepts for CVID are sufficient to treat these patients. Stem cell transplantation is a therapeutic option that must be considered. However, owing to lack of prospective data on the natural history of this form of CID, the indications remain unclear.
In view of the above considerations, there is need for a study providing data on the natural history and prognosis for patients with P-CID, in particular with respect to the long-term outcome with and without SCT. In addition, a platform for molecular and immunological characterization for patients with P-CID, which helps in the establishment of molecular diagnosis including the elucidation of novel genetic defects, is needed. On the basis of these data, it will be possible to make more balanced treatment decisions, in particular with respect to stem cell transplantation in this heterogeneous patient group.
Hematopoietic stem cell transplantation as therapy for combined immunodeficiencies (2.1.2)
In patients with SCID, hematopoietic stem cell transplantation (HSCT) represents the standard treatment, and significant experience with this form of therapy has been documented (Buckley 2000b; Myers et al. 2002; Grunebaum et al. 2006; Mazzolari et al. 2007; Griffith et al. 2008; Railey et al. 2009). The ideal source of allogeneic stem cells is an HLA-matched sibling. In patients lacking a histocompatible sibling donor, either parental or unrelated stem cell donors have been used. For SCID patients with an HLA-matched sibling donor, the widely used approach is not to give conditioning prior to stem cell transplantation. Conditioning regimes of varying intensity are used for patients without an HLA-matched sibling donor (Grunebaum et al. 2006; Mazzolari et al. 2007; Griffith et al. 2008).
Using these approaches, the majority of SCID patients will have reconstitution of T-cell function that is immunologically protective. The vast majority, if not all, of the T cells will be of donor origin; in contrast, the extent of donor cell engraftment in B, NK, myeloid, and erythroid lineages will be variable (Haddad et al. 1999; Neven et al. 2009). Many patients require chronic immunoglobulin replacement therapy. Despite these limitations, the survival rate of transplanted SCID patients is above 70% in most large centres (Mazzolari et al. 2007; Neven et al. 2009). However, some recent studies addressing the long-term clinical and immunologic outcomes of patients with SCID have shown that up to half of the patients followed long-term post-HSCT experience one or more significant clinical events, including persistent chronic graft-versus-host disease (GVHD), autoimmune and inflammatory manifestations, opportunistic and nonopportunistic infections, chronic HPV warts, and a requirement for nutritional support (Patel et al. 2008; Railey et al. 2009; Neven et al. 2009; Sarzotti-Kelsoe et al. 2009)).
There is much less documented experience with HSCT for patients with P-CID. In contrast to SCID patients, P-CID patients have significant numbers of circulating T cells with variable residual function. These cells may interfere with proper engraftment and may complicate the procedure of HSCT. Conditioning is therefore usually also used with matched sibling donors, but the intensity is highly variable. Whether some of the principles learned from HSCT for SCID can also be applied to P-CID remains open. Particularly, in older patients with P-CID, the risk of the transplantation procedure must be even more carefully balanced against its benefits. Reconstitution of B-cell immunity is a more relevant issue in P-CID, and it will be an important task to identify factors for reconstituting B-cell immunity with minimal toxicity.
Overall, the inconsistent treatment approaches to P-CID and the limited clinical reporting to date emphasize the critical need for a prospective multicentre study of P-CID treatment. Even though various protocols of HSCT are employed, each enrolled patient will be evaluated using a uniform set of documentation items. As mentioned above, it will be of similar importance to follow-up with patients who were not transplanted with the same set of parameters. The P-CID study will thus provide an unprecedented opportunity for systematic long-term follow-up of patients with P-CID throughout Europe.
Own previous work (2.2)
Combined immunodeficiencies have been an interest of our group for the last 5 years. In an attempt to understand the molecular basis of these disorders, we have carefully evaluated the clinical, immunological, and genetic basis in several individual patients. Thus, we have described the clinical and immunological consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency (Speckmann et al. 2008) and we have characterized the clinical phenotype and the immunological consequences of hypomorphic mutations in several genes involved in V(D)J recombination (Ehl et al. 2005; Enders et al. 2006; Rohr et al. 2010). Recently, we have also identified ADA deficiency in 2 patients with a late-onset CID (Speckmann et al. 2012). Overall, these patient observations significantly contributed to the phenotypic description of the heterogeneous immunodeficiencies caused by hypomorphic mutations in genes usually associated with SCID. We have put this own previous work into a larger perspective in the form of a literature review on atypical SCID, where we identified 73 patients with mutations in SCID-causing genes, T cells above 100/µL and a presentation different from SCID and Omenn syndrome (Felgentreff et al.). The mean age of onset of significant clinical symptoms was 1.8 (0–17) years and the mean age at diagnosis was 8 (0–39) years. Before diagnosis, 5 of the patients never had a severe infection, whereas 35 had at least 1 feature of immune dysregulation, including autoimmune cytopenia, lymphoproliferation, granulomas, or chronic diarrhoea. Autoantibody-mediated disease almost exclusively occurred in B-SCID variants. Lymphopenia (< 1000/µL) was noted in 21 of 40 patients, but 13 had normal T-cell counts. Naïve CD45RA+ CD4+ T cells were below 20% in 15 of 22 patients. Ten of 36 patients had normal IgG and 8 of 26 had protective titers to at least one vaccine. Twenty-one of 35 patients had elevated IgE, and 16 of these patients had adenosine deaminase (ADA) mutations. Overall, the findings of this overview characterize atypical SCID as a distinct form of P-CID with immune dysregulation in addition to infection susceptibility. The clinical and immunological data provide an important basis for the design of the P-CID study, which will include a significant number of patients with atypical SCID.
In addition to these scientific contributions, we have set up the infrastructure for such an international trial at the Centre of Chronic Immunodeficiency (CCI) within the last 2 years. The Advanced Diagnostic Unit at the CCI is well equipped for cutting edge studies on T-cell immunity and in constant exchange with the experimental groups at the CCI. One of the research foci of the CCI is on T-cell immunity. A clinical study unit has been set up and epidemiological and biostatistical support is available. Finally, animal models for CIDs have been set up with a focus on infections and immune dysregulation. Based on the BMBF-funded PID-Net, the CCI is part of a closely collaborating network of immunodeficiency centres in Germany and this will favour effective patient recruitment.
The groups involved in the P-CID study include 20 major centres with long-established immunology and transplant programs engaged in research and therapy of primary immunodeficiencies. These centres have individually contributed enormously to our current knowledge of the genetics, pathogenesis, and treatment of CIDs
Rationale and objective
Rationale (3.1)
We postulate that the nature and the extent of the T-cell deficiency and their development over time are key factors determining the prognosis for CID patients that are to some extent independent of the particular molecular diagnosis. We argue that in addition to factors specific for certain molecular causes of CID such as radiosensitivity or syndromal manifestations, knowledge about the association of the degree of impairment of T-cell immunity with outcome (mortality, morbidity, quality of life (QOL)) will be helpful in making appropriate decisions on whether and when to transplant CID patients. We think that consideration of CID as a group of diseases is justified in this context, because many molecular diagnoses associated with CID are so rare that it is highly unlikely that treatment recommendations will be derived from larger cohort studies. In this study, we focus on children in whom HSCT is a relevant consideration at study entry, i.e., patients who have a documented T-cell deficiency that is profound enough to have caused a significant clinical manifestation such as a severe infection or a severe manifestation of impaired immune regulation. For the purpose of this study, these patients are classified as suffering from profound combined immunodeficiency (P-CID).
A prospective multicentre study of patients with P-CID has not been performed previously. The long-term outcome of patients with this condition therefore remains poorly characterized, and the appropriate treatment as well as the appropriate time point and criteria for interventions such as HSCT remain undetermined. This natural history study includes nontransplanted patients with P-CID who are treated according to centre policy by participating institutions. To determine the clinical and laboratory patient variables that are most important in determining outcome, we will uniformly collect clinical data and biomarkers on all patients entered into this study. At study entry, there will be a retrospective documentation of severe infections, severe immune dysregulation, and malignancies (severe events) and such events and survival will then be documented prospectively throughout the 5-year observation period.
Patients will be distributed among 3 strata: (i) patients for whom the decision to transplant is made at the time point of study entry (group 1, primary HSCT); (ii) patients who are initially followed without transplantation, but for whom a decision to transplant is made during the study period (group 2, secondary HSCT); and (iii) patients, who are not transplanted during the observation period (group 3, no HSCT). Patients receiving alternative therapies, in particular PEG-ADA or gene therapy, will be followed according to treatment-specific study protocols.
The American Primary Immune Deficiency Treatment Consortium (PIDTC) has recently conceived a Prospective Natural History Study of Diagnosis, Treatment, and Outcomes of Children with SCID Disorders (RDCRN PIDTC Protocol # 6901). That study concentrates mainly on patients with SCID (who are excluded in the P-CID study), but also includes patients with leaky SCID and Omenn syndrome. Although all patients in the U.S. study undergo HSCT at study entry, only a part of the patients in the P-CID study will be transplanted initially. Nevertheless, there is a significant patient group that would be eligible for the U.S. and the European study. It is our intention to collaborate with this U.S. initiative and to provide a data framework in the P-CID study that will allow comparison of the data obtained in the 2 studies. For this, all patients transplanted in the P-CID study will be entered into the stem cell transplantation for immunodeficiencies (SCETIDE) registry.
On the basis of this HSCT documentation, we aim to identify variables contributing to the best outcomes for HSCT, which is an important therapeutic option for patients with P-CID. Because P-CID includes a spectrum of immunologic presentations, and there have been several approaches to transplant, there are many questions of interest and variables to be explored. All patients undergoing primary or secondary HSCT will be included in this analysis.
The results of this study will help to make treatment decisions for patients with non-SCID T-cell deficiencies. In particular, it will provide information about the risks and benefits of HSCT in P-CID patients and about the time point when HSCT is indicated. In collaboration and extension of the U.S. study, our investigations will also provide data that defines optimal conditions for the achievement of immune reconstitution. The results will also shape future directions for trials of HSCT and other cellular therapies applied to P-CID patients.
Objectives
Study objectives, all P-CID patients (3.2.1)
The primary objective of the study is to provide natural history data on patients with P-CID, irrespective of whether they undergo HSCT or not. The goal is to determine survival 5 years after study inclusion. The event analysed is death from any cause. The time to this event is the time from the first major infection or major manifestation of immune dysregulation or malignancy (documented retrospectively at the time of diagnosis) to death. As a secondary endpoint we analyse the time point of HSCT. The time to this event is the time from the first major infection or major manifestation of immune dysregulation or malignancy to HSCT. A tertiary endpoint is the frequency of severe events (major infections, major manifestations of immune dysregulation, malignancy, and major transplant-related complications) during the observation period. In addition, we will determine QOL at each study visit during the observation period.
The secondary objective is to develop a risk model for P-CID patients. The model considers severe events and potentially predictive biomarkers. Events prior to study inclusion are summarized in a morbidity score that is obtained at study inclusion and that is weighted for the time of exposure (i.e., chronological age and time of first severe event). In the prospective part of the study, severe events and potentially predictive biomarkers are obtained yearly for an observation period of 5 years. Major clinical patient characteristics and biomarkers will then be used as prognostic factors in survival models to establish the risk model.
Study objectives, HSCT patients (3.2.2)
The primary objectives of this part of the study are to estimate the 6 month and 1 year overall survival probabilities for patients after HSCT for P-CID and to study risk factors for overall survival in this group of transplanted patients. A variety of patient, donor, and transplant factors will be evaluated for their contribution to the outcomes described above.
The secondary objectives of this part of the study are to determine the effects of donor, recipient, and treatment factors on the proportion of subjects having durable engraftment within each blood lineage, and the proportion of subjects having successful or sustained immunologic reconstitution, including T-cell function and B-cell function, after allogeneic HSCT for CID. Additionally, we will evaluate donor, recipient, and transplant factors as well as engraftment and quality of immune reconstitution as contributors to clinical outcome; for example, occurrence of post-transplant infections, GVHD, autoimmunity, growth and development, and QOL.
Objectives for patients who receive alternative therapies (3.2.3)
A small group will include patients who receive alternative therapy; i.e., those with ADA deficiency who are treated with PEG-ADA or patients with ADA deficiency or XSCID variants undergoing gene therapy. As these alternative forms of therapy are mostly used for patients with SCID and not P-CID, this will represent a relatively small group of patients. However, the study will be open to these patients to provide standardized and comparable follow-up data.
Methods
Endpoints: The primary endpoint is overall survival determined after year 5. The event analysed is death from any cause. The time to this event is the time from the first major infection or major manifestation of immune dysregulation (documented retrospectively at the time of study entry) to death. The secondary endpoint is the time point of HSCT. The time to this event is the time from the first major infection or major manifestation of immune dysregulation to HSCT. The tertiary endpoint is the frequency of major infections or major manifestations of immune dysregulation during the observation period. These endpoints will be used as prognostic factors in combination with a set of potentially predictive biomarkers in survival models to establish a risk model for P-CID patients.
In addition, within this study, all patients undergoing HSCT will be analysed with a second set of endpoints to evaluate of the outcome of SCT: The primary endpoint is overall survival after 6 months and 1 year of follow-up. The secondary endpoints are engraftment, immune reconstitution, and clinical outcome assessed at 6 and 12 months after HSCT.
TIMETABLEStart of Study: July 2011End of the Study: June 2021
Sample size: We assume that 40 patients per year (a total of 200 patients) will be included. Of these, it is estimated that 40% (80 patients) will receive primary HSCT, 30% (60 patients) secondary HSCT, and 30% (60 patients) no HSCT. The main trial objectives can also be achieved, if fewer patients are recruited.
The characteristics of the patient cohort (presence and variability of molecular diagnosis, variability in decision to transplant) are difficult to foresee. Evaluation of a pilot phase (until June 2014) will provide this information. It will then be evaluated to which degree patients can be matched by the morbidity score obtained at study entry for the outcome studies. To determine how many patients will be needed to compare outcomes in those undergoing 1° versus 2° versus no HSCT, sample size calculations have been performed. Assuming a reference 5-year mortality of 20%, 60 patients in each of the 3 groups will allow us to detect a relative risk of 2.2 with a power of 80%.
Statistical analysis: The risk and prognostic factors for the need of HSCT and the impact of HSCT on death will be analysed with survival analysis using multistate models. Left-truncation (delayed entry) will be addressed. Appropriate regression models will be applied.
The impact of HSCT on the frequency of severe events (infections, severe manifestations of immune dysregulation, transplant-related complications) will be examined using models to analyse recurrent events.
Inclusion criteria: Clinical and immunological criteria determine inclusion irrespective of the genetic diagnosis.
T-cell criteria (2 out of 4)
•
Reduced T-cell counts (CD4: < 700, if < 2 years of age; < 500, if 2–4 years of age; < 300, if > 4 years of age; CD8: < 350, if < 2 years of age; < 250, if 2–4 years of age; < 150, if > 4 years of age)
•
Reduced thymic function (CD45RA+CD62L+ or CD45RA+CD31+ of CD4+ < 30% < 2 years of age, < 25% 2–6 years of age, < 20% > 6 years of age)
•
Impaired T-cell proliferation (PHA or anti CD3/CD28 response < 30% of lower limit of normal)
•
Elevated fraction of γ/δ T cells (> 15% of total CD3+ T cells)
AND Clinical criteria
•
At least 1 major infection criteria (viral, bacterial, opportunistic); or
•
At least 1 major immune dysregulation criteria (granulomas, lymphoproliferative disease, unexplained interstitial lung disease, inflammatory bowel disease, autoantibody mediated disease, vasculitis); or
•
At least 1 malignancy criteria (lymphoid malignancies and virally induced malignancies)
AND age > = 1 year and < = 16 year at study inclusion
Exclusion criteria:
•
No written informed consent from patient, or parents in case of minors, available or no assent of minor if applicable
•
Patients with a clinical diagnosis of SCID or Omenn syndrome within the first year of life
•
P-CID Patients for whom decision for HSCT is taken at age < 1 year
•
Patients with Wiskott–Aldrich syndrome and CD40 Ligand Deficiency, because disease-specific prognosis and treatment data are available
•
Patients undergoing gene therapy or ADA enzyme replacement will be followed using the same parameters, but will not be included in the analysis.
Study plan
Study design (4.1)
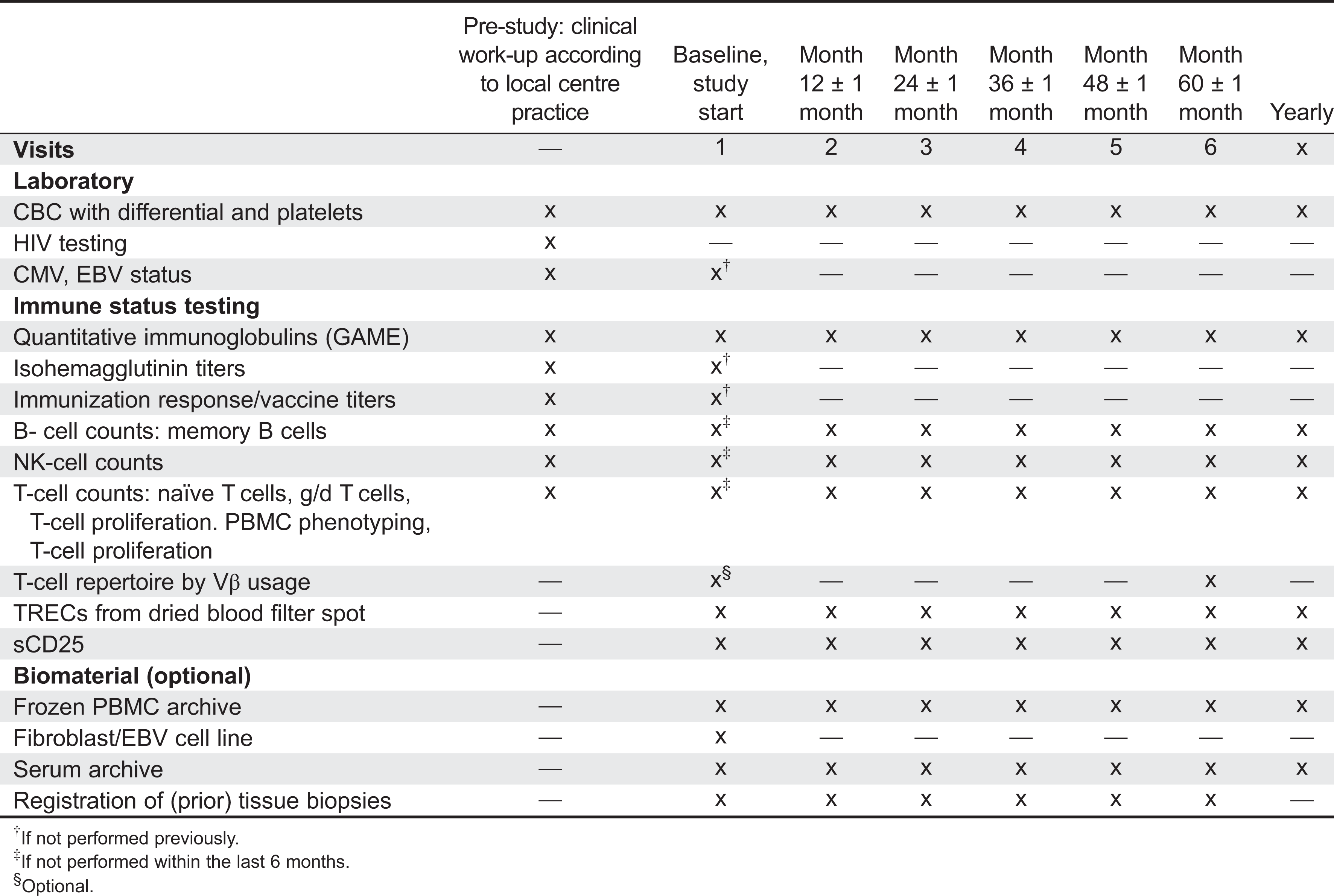
The study is conceived as a prospective outcome study and is summarized in Figure 1 and Tables 1 and 2. At enrolment, all major infections, major manifestations of immune dysregulation, and malignancies (severe events) that have occurred prior to study entry, are documented. Furthermore, the time point and the immunological laboratory characteristics leading to the initial diagnosis of P-CID are noted. From these data, a baseline morbidity score is determined that considers the time of exposure (both age and time after first severe event). Additional baseline immunological laboratory diagnostics is requested, if the last investigations have been performed more than 6 months prior to study entry.
Figure 1:
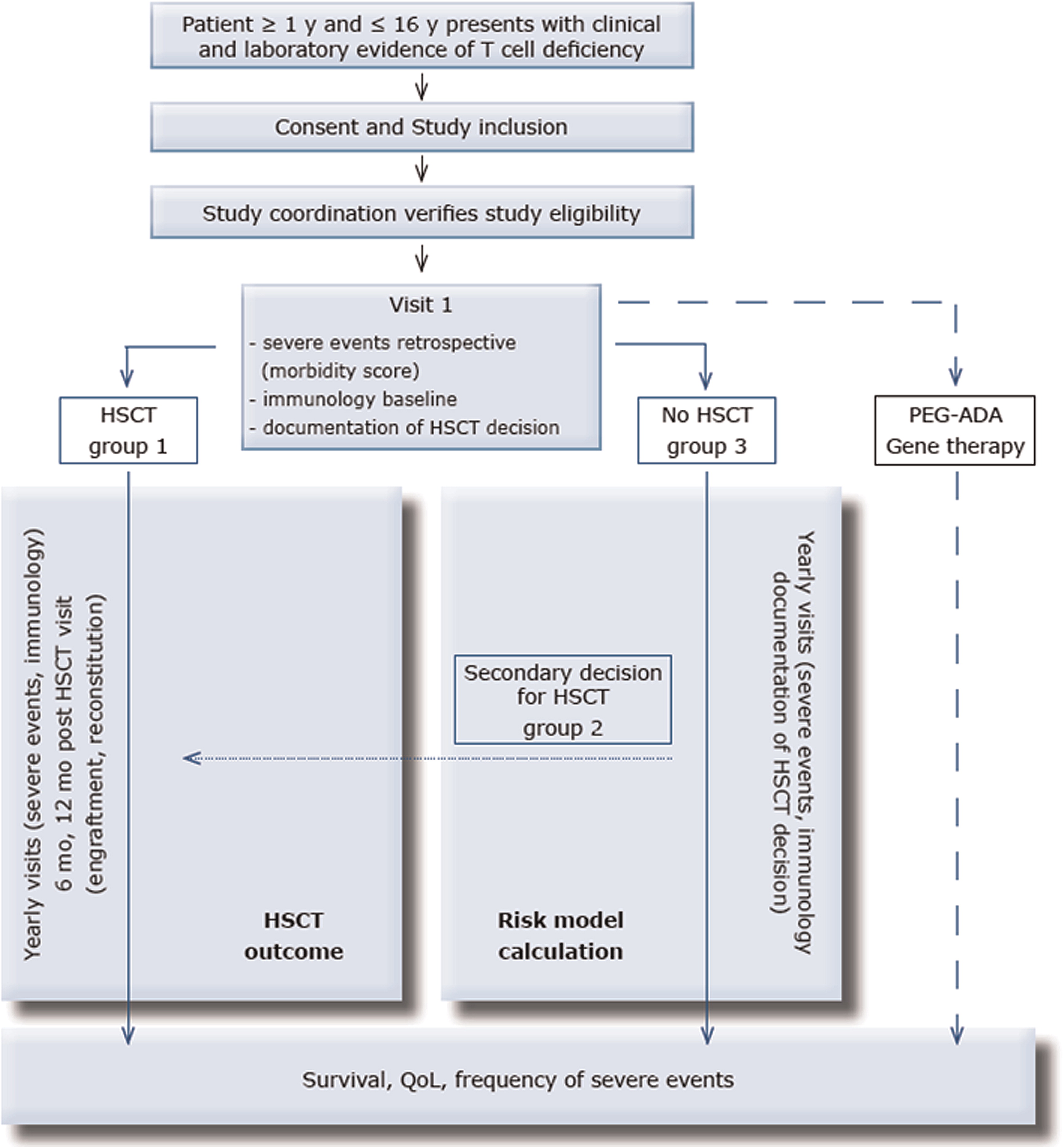
Table 1:
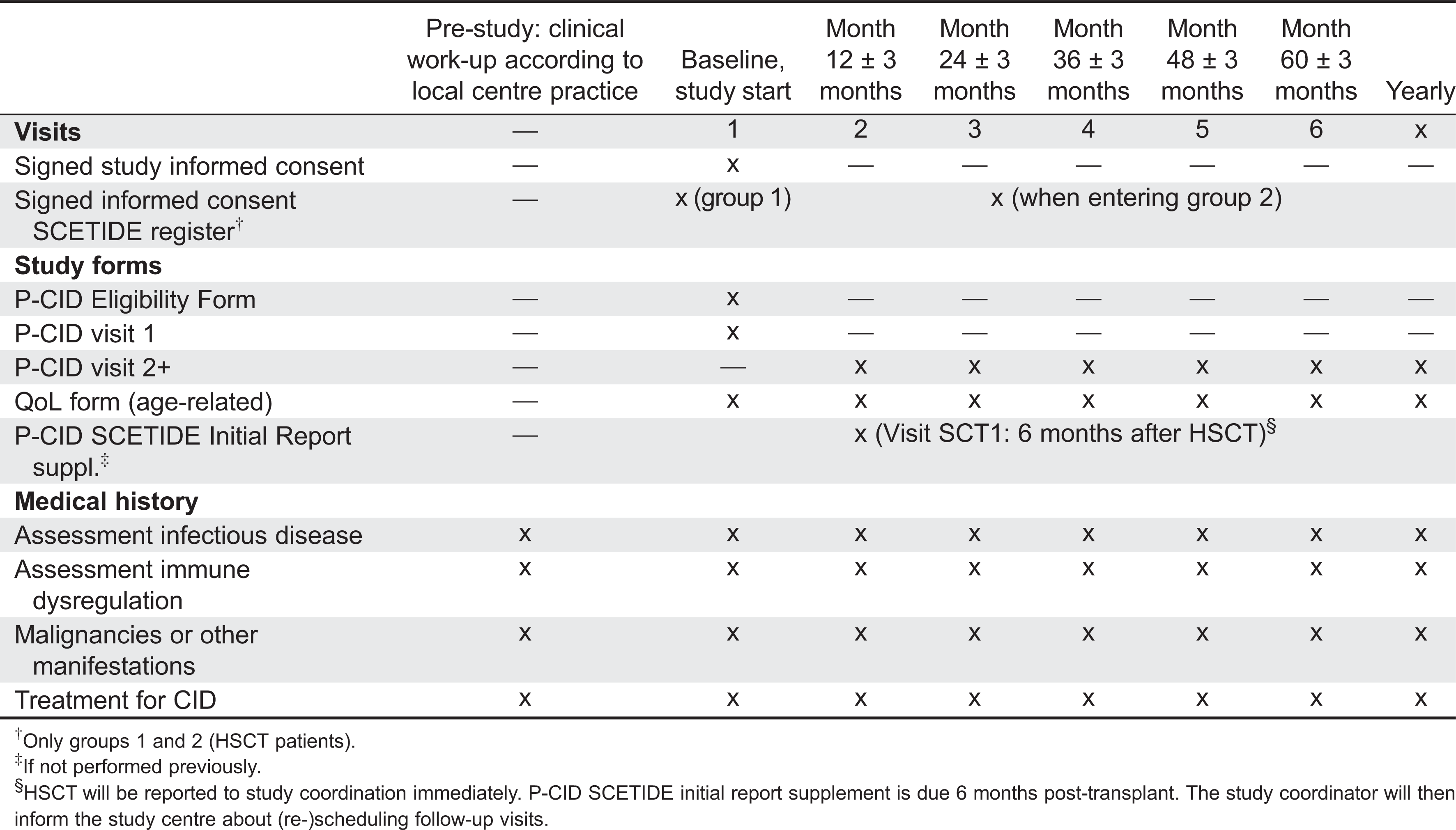
Table 2:
Patients then either undergo primary HSCT within 1 year after enrolment (group 1), secondary HSCT within the further follow-up period (group 2), or they are not transplanted during the observation period (group 3). Few patients will receive alternative therapies (enzyme replacement therapy, ERT; gene therapy, GT). The individual treatment decision is based on policies of the local centre. Arguments leading to the decision for or against HSCT will be recorded in a standardized fashion; this information will be obtained yearly for those patients remaining without transplant. After entry into the study, all patients will be followed up yearly for 5 years. All patients will receive yearly follow-up visits from the time point of study entry until the end of the study. Patients undergoing 2° HSCT will undergo an additional study visit 6 months after HSCT and the following visits will be rescheduled to occur yearly after transplantation (instead of yearly after study inclusion.
All patients will be monitored for survival, infections, manifestations of immune dysregulation, malignancy, QOL, and predictive biomarkers. Patients undergoing HSCT will in addition be monitored for engraftment, immune reconstitution, and a set of clinical parameters and biomarkers related to HSCT. All severe events (severe infections, severe manifestations of immune dysregulation, malignancy, severe transplant-related complications) occurring during the observation period are documented. The time from the first severe event (documented retrospectively at the time of study entry) to death will be determined and compared in the 3 patient groups. Furthermore, the time from the first severe event to HSCT will be determined. From these clinical data and from potentially predictive biomarkers determined at diagnosis, study entry and yearly thereafter, a risk model will be calculated for patients with P-CID. Finally, the frequency of severe events and QOL during the follow-up period will be determined in the 3 groups irrespective of whether they are transplanted or not.
In addition, all patients undergoing HSCT will be analysed with a second set of endpoints to evaluate of the outcome of HSCT. Engraftment and immune reconstitution will be assessed at 6 and 12 months after HSCT. The clinical outcome based on follow-up data obtained at 6 and 12 months will be determined.
Study endpoints for all P-CID patients
Primary endpoint (4.2.1)
The primary endpoint is overall survival determined after 5 years of follow-up. The event analysed is death from any cause. The time to this event is the time from the first major infection, major manifestation of immune dysregulation, or malignancy (documented retrospectively at the time of diagnosis) to death.
Definition of major infections:
•
Opportunistic infection (pneumocystis, invasive NTM, invasive fungal infection, oesophageal candidiasis, persistent cryptosporidiosis, other)
•
Severe bacterial infection (necessitating PICU)
•
Recurrent pneumonia leading to chronic lung disease (e.g. bronchiectasis)
•
Invasive bacterial infection (sepsis, meningitis, OM, septic arthritis, purulent pericarditis, mastoiditis, complicated pneumonia, abscess except skin)
•
Severe viral infection (requiring PICU)
•
Persistent viremia (CMV, EBV, Adeno)
•
Persistent severe viral skin lesions
•
Other severe infections
Definition of major manifestations of immune dysregulation:
•
Biopsy-proven granulomatous disease
•
Inflammatory bowel disease
•
Unexplained interstitial lung disease
•
Autoimmune cytopenia or autoimmune endocrinopathy
•
Lymphoproliferation (persistent splenomegaly or lymphadenopathy)
•
Systemic vasculitis (more than skin involvement)
•
Other severe manifestations of immune dysregulation
Other major manifestations:
•
Malignant disease
•
Other major disease manifestations (requiring hospitalization) related to P-CID
Secondary endpoint (4.2.2)
The secondary endpoint is the time point of HSCT. The time to this event is the time from the first major infection or major manifestation of immune dysregulation to HSCT.
Tertiary endpoint (4.2.3)
Tertiary endpoint is the frequency of severe events (major infections, major manifestations of immune dysregulation, malignancy or major transplant-related complications) during the 5 year observation period. Major transplant-related complications regarded as severe events are defined below (4.3.2.3). Patients will be evaluated yearly and the time point of all severe events will be documented.
Quality of life will be evaluated at study entry and yearly thereafter using the Peds QL Generic Core Scales (Child Self Report and (or) Parent proxy Report, depending on recipient's age). Transplanted patients will receive both the Generic Core Scale as well as the Peds QL Transplant Module.
Potentially predictive biomarkers (4.2.4)
To develop the risk model, biomarkers obtained at diagnosis of P-CID (retrospective documentation), at study entry (if more than 6 months later than diagnosis), and at the yearly follow-up visits will be considered. The following biomarkers will be obtained:
T cell
•
CD3
•
CD4, CD8
•
CD4 naïve (CD45RA+)
•
Activated CD4, CD8 (HLA-DR)
•
T cells expressing γ/δ TCR
•
T-cell proliferation to PHA and aCD3
•
TRECs and KRECs (central analysis of dried blood spots from Guthrie cards; see below)
•
Optional: T-cell diversity (V-beta usage)
B cell
•
IgA, IgM levels; IgG when off Ig supplementation
•
Specific antibodies and Isohemagglutinin levels (determined once)
•
Naïve B cells
•
CD19+ CD27+ Memory B cells
Other
•
NK cells
•
sCD25
Additional study endpoints for the patients undergoing HSCT
Primary endpoint, transplanted patients (4.3.1)
The primary endpoint is overall survival after 6 months and 1 year of follow-up. The event analysed is death from any cause. The time to this event is the time from HSCT to death or last follow-up.
Secondary endpoints, transplanted patients (4.3.2)
Immune Reconstitution
Immune reconstitution is defined separately for T cells and B cells based on attainment of lab test values at pre-specified time points.
Full T-cell reconstitution (6 and 12 months post-HSCT)
•
Lymphocyte proliferation to PHA and aCD3 > 30% of lower limit of normal control, and
•
Donor T cell chimerism ≥ 80%
Full B-cell reconstitution (12 months post-HSCT)
•
Four-fold increase in anti-tetanus antibody following tetanus immunization (without replacement Ig administration during the testing period) OR
•
Four-fold increase in another specific antibody response following vaccination (without replacement Ig administration during the testing period)
Engraftment
For whole blood and subsets the following criteria will be used at 6 and 12 months as endpoints: < 5% donor = autologous reconstitution; 5–80% donor = mixed chimerism; ≥ 80% donor = full chimerism.
•
Whole blood
•
CD3
•
CD19
•
CD14 and (or) CD15 (myeloid cells)
•
CD3– CD56+ NK cells
Graft failure/rejection is defined as:
•
Less than 5% of donor CD3 cells at 6 months using standard PCR-based or in situ hybridization techniques, or
•
Second transplant (unless > 5% CD3 and purpose is to boost immune recovery)
Clinical Outcome
Clinical outcome will be evaluated for the occurrence and resolution of pre- and post-transplant infections, acute and chronic GVHD, development of autoimmunity, retardation of growth or development, and QOL.
Infections
Incidence of infection post-HSCT. These will be reported by site of disease, organism, date of onset, and resolution, if any, at 6 and 12 months post-HSCT.
Growth and Nutrition
•
Weight and height pre-HSCT and 1 year post-HSCT
•
Chronic diarrhea, and (or) requirement for supplemental nutrition including tube feeding or TPN
GVHD
•
Occurrence of acute (grade II–IV and grade III–IV) GVHD at 6 months post-HSCT. Any skin, gastrointestinal, or liver abnormalities fulfilling the consensus criteria of grade II–IV acute GVHD or grade III–IV acute GVHD are considered severe events. Death is a competing risk, and patients alive and without acute GVHD will be censored at the time of last follow-up
•
Occurrence of chronic GVHD at 6 and 12 months post-HSCT. Occurrence of symptoms in any organ system fulfilling the criteria of limited or extensive chronic GVHD. Death is a competing risk, and patients alive and without chronic GVHD will be censored at time of last follow-up
•
Therapy given for GVHD
•
Time to discontinuation of immunosuppression
Autoimmunity
Occurrence of autoimmunity requiring treatment with immunosuppression or other therapy. Death is a competing risk, and patients alive and without autoimmunity will be censored at time of last follow-up. Date of onset and type of treatment will be collected on the following:
•
Autoimmune hypothyroidism
•
Autoimmune cytopenia (hemolytic anemia, thrombocytopenia, neutropenia)
•
Arthritis
•
Myositis
•
Nephritis
•
Bronchiolitis obliterans or other pulmonary autoimmune disease
•
Vitiligo
•
Alopecia
•
Inflammatory bowel disease
•
Neurodegeneration
•
Vasculitis
Other complications of HSCT needing treatment
•
Veno-occlusive disease
•
Thrombotic thrombocytopenic purpura
•
Bronchiolitis obliterans / chronic lung disease
•
Seizures
•
Hypertension
•
Malignancy
•
Other
Quality of Life
For transplanted patients, a separate, adapated QOL form (Peds QL Transplant Module) is available. For transplanted patients, QOL testing will be done using both the Peds QL Generic Core Scales (Child Self Report and (or) Parent proxy Report, depending on recipient's age) and the Peds QL Transplant Module.
Consideration of bias
A number of factors can influence the outcome for patients recruited in this study. Characterization of variables predicting outcome will have to address these factors (see statistical analysis).
Heterogeneity of diagnoses: The molecular diagnoses of the patients will be heterogenous, leading to an immunological and clinical variability in the patient cohort. Immunological variability will be addressed by grouping the patients into 4 subgroups: patients with reduced T-cell numbers, but normal proliferative response (+/−) intrinsic B-cell defects; and patients with normal or reduced T-cell numbers, but reduced Tcell proliferation (+/−) intrinsic B-cell defects. It will be determined whether these variables contribute to outcome.
Heterogeneity of age at study inclusion: The age at study inclusion will be between 1 and 16 years of age, and therefore the time of exposure to the risks of the T-cell deficiency will be variable. The statistical analysis will address this problem by left-truncation to account for delayed entry.
Heterogeneity of history before study inclusion: The patients will have had a different profile of severe events (infections, manifestations of immune dysregulation, malignancy) prior to study inclusion and different degrees of CID-related chronic organ pathology (e.g., chronic lung disease). This problem will be accounted for by assessing morbidity for acute and chronic severe events, which will be retrospectively determined at study entry. After the observation period, patients can then be compared on the basis of their morbidity profile at study entry.
Centre effect and treatment heterogeneity: It is expected that centre policy will have an important impact on the decision to transplant. Centre experience, including the number of transplantations performed and the personal experience of the transplanting physician, will have an impact on the recommendation to the patient. Additional patient-specific variables include the donor availability and the will of the patient's family. This heterogeneity can be used to identify matched pairs of patients with a similar morbidity profile but different decisions on HSCT.
Treatment period effect: Owing to the long recruitment and observation period (covering more than 10 years in total), it is expected that a treatment period effect needs to be considered. Patient management may change because of the introduction of new drugs (e.g., antivirals, immunosuppressive drugs) with an impact on the significance of certain clinical manifestations on the prognosis. This problem will be addressed by introducing a period variable (such as corresponding year in calendar time for each patient) in the model.
Timetable (4.4)
•
Enrolment of first subject: Q3/2011
•
Evaluation of pilot phase: Q1/2015
•
Enrolment of last subject (planned): Q3/2017
•
Last visit of last subject (planned): Q3/2022
•
Completion of statistical analysis (planned): Q1/2023
•
Contacts per subject: yearly (according to local practice)
•
Planned duration of follow-up: 5 years per subject
Participating centres (4.5)
About 25, mostly European, centres (Germany, Austria, Italy, United Kingdom, France, Spain, Czech Republic, Slovenia, Switzerland, Kuwait, Israel, and Canada) have planned to participate. All participating centres meet the structural and personnel requirements for performing the planned regular study-related investigations.
Number of study participants (4.6)
Two hundred patients will be enrolled in the study. A feasibility assessment has been performed. Feasibility will be reassessed after evaluation of the pilot phase in Q1/2015.
Subject population and selection criteria
Inclusion criteria (5.1)
Patients will be proposed for enrolment in this natural history study by investigators at participating institutions; after consent has been obtained, basic data on T-cell parameters and prior severe events will be submitted for review (see below) to determine study eligibility.
The following criteria have to be met by a patient to fulfil the P-CID case definition.
•
T-cell criteria (2 of the following: reduced CD4+ or CD8+ T cell counts, reduced fraction of naïve CD4 T cells, increased fraction of γ/δ T cells, impaired proliferation to PHA, or anti-CD3) AND
•
At least 1 major infection criterion (viral, bacterial, opportunistic), or
•
At least 1 major immune dysregulation criterion (granulomas, lymphoproliferative disease, unexplained interstitial lung disease, inflammatory bowel disease, autoantibody mediated disease, vasculitis), or
•
At least 1 malignancy criterion (lymphoid malignancies and virally induced malignancies)
•
Age > = 1 year and < = 16 years at study inclusion
Definition of T -cell criteria:
•
Reduced T-cell counts (CD4: < 700, if < 2 years of age; < 500, if 2–4 years of age; < 300 if > 4 years of age; CD8: < 350, if < 2 years of age; < 250, if 2–4 years of age; < 150, if > 4 years of age)
•
Reduced fraction of naïve CD4+ T cells (CD45RA+CD62L+ or CD45RA+CD31+ of CD4+ < 30% < 2 years of age, < 25% 2–6 years of age, < 20% > 6 years of age)
•
Elevated fraction of γ/δ T cells (> 15% of total CD3+ T cells)
•
Impaired T-cell proliferation (PHA or anti CD3/CD28 response < 30% of lower limit of normal)
The following molecularly defined diseases are eligible for inclusion, provided they fulfil the entry criteria. Some of these diseases will only fulfil the entry criteria in rare variants. The rationale for choosing such a broad approach is that the clinical problem concerning the decision for indication and time point of HSCT is similar in all of these patient groups. In all patients the clinical consequences of T-cell deficiency is a major factor limiting survival. It is acknowledged that for some subgroups (e.g., radiosensitive disorders), additional specific risk factors such as malignancies have an important impact on survival. Similarly, neurodevelopmental problems associated with some forms of P-CID can have a significant impact on QOL. Therefore, subgroup analysis should be performed for some diseases, which will be limited by patient numbers. Overall, however, a significant number of core diseases will remain. Moreover, the value of a common standardized documentation platform for the study of the natural history of this poorly studied group of CIDs is rated higher than these.
Group 1: CID
•
BCL10 deficiency
•
CARD11 deficiency
•
Cartilage hair hypoplasia
•
Caspase 8 deficiency
•
CD25 deficiency
•
CD27 deficiency
•
CD3γ deficiency
•
CD8 deficiency
•
Coronin-1A deficiency
•
CTPS1 deficiency
•
DOCK8 deficiency
•
Dyskeratosis congenita
•
ITK deficiency
•
ICF syndrome (types 1 and 2)
•
LCK deficiency
•
MAGT1 deficiency
•
MALT1 deficiency
•
MHC I deficiency
•
Moesin deficiency
•
OX40 deficiency
•
PNP deficiency
•
RHOH deficiency
•
RLTPR deficiency
•
STIM1 deficiency
•
STAT5b deficiency
•
Schimke syndrome
•
STK4 (MST1) deficiency
•
TPP2 deficiency
•
TRAC deficiency
•
UNC119 deficiency
•
ZAP70 deficiency
Group 2: CIDs with radiosensitivity
•
Radiosensitive SCID variants
•
Cernunnos deficiency
•
Nijmegen Breakage syndrome
Group 3: Immunodeficiencies associated with a CID phenotype in some cases
•
All genetic diseases associated with SCID (> 20 molecularly defined entities)
•
LRBA deficiency
•
PI3K delta activating mutations
•
PI3K p85 deficiency
•
CTLA4 deficiency
•
STAT1 activating mutations
•
STAT3 activating mutations
•
Atypical late-onset FHL (MUNC13, MUNC18, STX11, Perforin)
•
MHC II deficiency
•
Atypical NEMO deficiency
•
Atypical IPEX
•
Atypical XLP (SAP/BIRC4)
•
WHIM syndrome
Patients with Wiskott–Aldrich syndrome, CD40 ligand deficiency, and ataxia teleangiectasia are excluded, owing to the existence of disease-specific data or registries addressing the questions of this study.
Exclusion criteria (5.2)
The following criteria will be used for exclusion:
•
No written informed consent from patient, or parents in case of minors, available
•
Patients fulfilling the clinical definition of SCID or Omenn syndrome before the first year of life
•
P-CID patients for whom decision for HSCT is taken at age < 1 year, because a clinical diagnosis of SCID or Omenn syndrome cannot be excluded
•
Patients with Wiskott–Aldrich syndrome CD40 ligand deficiency and ataxia teleangiectasia, because disease-specific prognosis and treatment data are available
•
Patients undergoing gene therapy or ADA enzyme replacement can be followed using the same parameters, but will not be included in the main analysis
•
Persons who are in a relationship of dependence or employment with the investigator
Enrolment of subjects in the study
Diagnostic workup prior to submission for eligibility (6.1)
At the local sites, as part of routine care and in advance of referral for the study, sufficient clinical assessment, immune phenotyping, and genetic testing (if available) will be performed to allow assignment of the diagnosis of P-CID. A list of available diagnostic and genetic facilities and their programs are provided on the P-CID website (www.pcid-study.org). Testing not available at the local sites may be performed on a research basis by the diagnostic laboratories of the participating centres. Molecular diagnosis is not required prior to eligibility procedures, although it may appropriately be performed locally as part of the diagnostic workup.
Information required for eligibility evaluation:
•
Medical history: infectious diseases, immune dysregulation, malignancy
•
Determination of CD4 and CD8 T cell counts
•
Determination of the fraction of naïve CD4+ T cells
•
Determination of the fraction of T cells expressing γ/δ TCR
•
In vitro T cell proliferation response to PHA
Informed consent (6.2)
After identifying a potentially eligible patient based on clinical and immunological criteria of P-CID, obtained by workup according to local centre practice, the investigator will inform the patient and the parents about the study and obtain informed consent. Informed consent forms (PICFs) can be downloaded from the CCI homepage (www.pcid-study.org). The signed original PICF will be filed in the patient's source documents, and a signed copy stays with the patient.
There is a section included in the P-CID PICF to inform the patient or parents that by consenting to the study participation, the consent for documentation of patient data into the SCETIDE registry is obtained. This allows data transfer to the SCETIDE registry if the decision is made to perform HSCT at any time during the study.
Eligibility procedures and registration (6.3)
Prior to final inclusion, all subjects must be approved by the P-CID Study Coordination. After having obtained informed consent, the local investigator will complete and submit the Study Eligibility Form, which can also be downloaded from the CCI homepage (see Section 7 in the Supplementary table of Contents1). This form does not include any personal patient data apart from date of birth (month and year). With the form, data on clinical history and immunological tests will be reviewed by the Medical Study Coordinator. If necessary, the Study Coordination will recontact the recruiting centre for further information. The evaluated Eligibility Form (Eligibility Evaluation Form) will be returned to the centre via email. For patients eligible for the study, the form will contain a patient identification number consisting of country code, a centre code, and the consecutive patient number. Both the Eligibility From and the Eligibility Evaluation Form are kept in the study folder at the recruiting site. All further forms (Case Report Forms, QOL Forms) will use the patient identification number and are thereby pseudonymised. Each centre will maintain a patient identification list in the study folder with the patient's name and the corresponding patient identification code to identify the patient anytime.
Footnote
1
Numbers following the heading correspond to the Table of Contents found in the Supplementary data available with the article through the journal Web site at http://lymphosign.com/doi/full/10.14785/lpsn-2015-0002.
REFERENCES
Allen R.C., Armitage R.J., Conley M.E., Rosenblatt H., Jenkins N.A., Copeland N.G., Bedell M., Edelhoff S., Disteche C., and Simoneaux D. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome Science. 1993 259 5097 990 -993
Andersen P., Borgan Ø., and Gill R. JSTOR: International Statistical Review / Revue Internationale de Statistique. 1982 50 3 219 -244
Buckley R.H. Primary immunodeficiency diseases due to defects in lymphocytes N. Engl. J. Med. 2000a 343 18 1313 -1324
Buckley R.H. Advances in the understanding and treatment of human severe combined immunodeficiency Immunol. Res. 2000b 22 2–3 237 -251
Coffey A.J., Brooksbank R.A., Brandau O., Oohashi T., Howell G.R., Bye J.M., Cahn A.P., Durham J., Heath P., Wray P., Pavitt R., Wilkinson J., Leversha M., Huckle E., Shaw-Smith C.J., Dunham A., Rhodes S., Schuster V., Porta G., Yin L., Serafini P., Sylla B., Zollo M., Franco B., Bolino A., Seri M., Lanyi A., Davis J.R., Webster D., Harris A., Lenoir G., De St Basile G., Jones A., Behloradsky B.H., Achatz H., Murken J., Fassler R., Sumegi J., Romeo G., Vaudin M., Ross M.T., Meindl A., and Bentley D.R. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene Nat. Genet. 1998 20 2 129 -135
Derry J.M., Ochs H.D., and Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome Cell. 1994 79 5 following 922
Ehl S., Schwarz K., Enders A., Duffner U., Pannicke U., Kuhr J., Mascart F., Schmitt-Graeff A., Niemeyer C., and Fisch P. A variant of SCID with specific immune responses and predominance of gamma delta T cells J. Clin. Invest. 2005 115 11 3140 -3148
Enders A., Fisch P., Schwarz K., Duffner U., Pannicke U., Nikolopoulos E., Peters A., Orlowska-Volk M., Schindler D., Friedrich W., Selle B., Niemeyer C., and Ehl S. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV J. Immunol. 2006 176 8 5060 -5068
Felgentreff K., Perez-Becker R., Speckmann C., Schwarz K., Kalwak K., Markelj G., Avcin T., Qasim W., Davies E.G., Niehues T., and Ehl S. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency Clin. Immunol. 2011 141 1 73 -82
Griffith L.M., Cowan M.J., Kohn D.B., Notarangelo L.D., Puck J.M., Schultz K.R., Buckley R.H., Eapen M., Kamani N.R., O'reilly R.J., Parkman R., Roifman C.M., Sullivan K.E., Filipovich A.H., Fleisher T.A., and Shearer W.T. Allogeneic hematopoietic cell transplantation for primary immune deficiency diseases: current status and critical needs J. Allergy Clin. Immunol. 2008 122 6 1087 -1096
Grunebaum E., Mazzolari E., Porta F., Dallera D., Atkinson A., Reid B., and Notarangelo L.D. Bone marrow transplantation for severe combined immune deficiency JAMA. 2006 295 5 508 -518
Haddad E., Le Deist F., Aucouturier P., Cavazzana-Calvo M., Blanche S., de Saint Basile G., and Fischer A. Long-term chimerism and B-cell function after bone marrow transplantation in patients with severe combined immunodeficiency with B cells: a single-center study of 22 patients Blood. 1999 94 8 2923 -2930
International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies, Notarangelo L.D., Fischer A., Geha R.S., Casanova J., Chapel H., Conley M.E., Cunningham-Rundles C., Etzioni A., Hammartröm L., Nonoyama S., Ochs H.D., Puck J., Roifman C., Seger R., and Wedgwood J. Primary immunodeficiencies: 2009 update J. Allergy Clin. Immunol. 2009 124 6 1161 -1178
Lin D.Y. Proportional means regression for censored medical costs Biometrics. 2000 56 3 775 -778
Liston A., Enders A., and Siggs O. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation Nat. Rev. Immunol. 2008 8 7 545 -558
Malphettes M., Gérard L., Carmagnat M., Mouillot G., Vince N., Boutboul D., Bérezné A., Nove-Josserand R., Lemoing V., Tetu L., Viallard J.-F., Bonnotte B., Pavic M., Haroche J., Larroche C., Brouet J.-C., Fermand J.-P., Rabian C., Fieschi C., and Oksenhendler E. Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect Clin. Infect. Dis. 2009 49 9 1329 -1338
Mazzolari E., Forino C., Guerci S., Imberti L., Lanfranchi A., Porta F., and Notarangelo L.D. Long-term immune reconstitution and clinical outcome after stem cell transplantation for severe T-cell immunodeficiency J. Allergy Clin. Immunol. 2007 120 4 892 -899
Myers L.A., Patel D.D., Puck J.M., and Buckley R.H. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival Blood. 2002 99 3 872 -878
Neven B., Leroy S., Decaluwe H., Le Deist F., Picard C., Moshous D., Mahlaoui N., Debre M., Casanova J.-L., Dal Cortivo L., Madec Y., Hacein-Bey-Abina S., De Saint Basile G., De Villartay J.-P., Blanche S., Cavazzana-Calvo M., and Fischer A. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency Blood. 2009 113 17 4114 -4124
Niehues T., Perez-Becker R., and Schuetz C. More than just SCID--the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2 Clin. Immunol. 2010 135 2 183 -192
Notarangelo L.D. Primary immunodeficiencies J. Allergy Clin. Immunol. 2010 125 2 Suppl 2 S182 -S194
Notarangelo L.D., Giliani S., Mazza C., Mella P., Savoldi G., Rodriguez-Perez C., Mazzolari E., Fiorini M., Duse M., Plebani A., Ugazio A.G., Vihinen M., Candotti F., and Schumacher R.F. Of genes and phenotypes: the immunological and molecular spectrum of combined immune deficiency. Defects of the gamma(c)-JAK3 signaling pathway as a model Immunol. Rev. 2000 178 39 -48
Patel N.C., Chinen J., Rosenblatt H.M., Hanson I.C., Brown B.S., Paul M.E., Abramson S.L., Ritz J., and Shearer W.T. Long-term outcomes of nonconditioned patients with severe combined immunodeficiency transplanted with HLA-identical or haploidentical bone marrow depleted of T cells with anti-CD6 mAb J. Allergy Clin. Immunol. 2008 122 6 1185 -1193
Pessach I., Walter J., and Notarangelo L.D. Recent advances in primary immunodeficiencies: identification of novel genetic defects and unanticipated phenotypes Pediatr. Res. 2009 65 5 Pt 2 3R -12R
Prentice R.L., Williams B.J., and Peterson A.V. On the regression analysis of multivariate failure time data Biometrika. 1981 68 2 373 -379
Railey M.D., Lokhnygina Y., and Buckley R.H. Long-term clinical outcome of patients with severe combined immunodeficiency who received related donor bone marrow transplants without pretransplant chemotherapy or post-transplant GVHD prophylaxis J. Pediatr. 2009 155 6 834 -840.e1
Rohr J., Pannicke U., Döring M., Schmitt-Graeff A., Wiech E., Busch A., Speckmann C., Müller I., Lang P., Handgretinger R., Fisch P., Schwarz K., and Ehl S. Chronic inflammatory bowel disease as key manifestation of atypical ARTEMIS deficiency J. Clin. Immunol. 2010 30 2 314 -320
Sarzotti-Kelsoe M., Win C.M., Parrott R.E., Cooney M., Moser B.K., Roberts J.L., Sempowski G.D., and Buckley R.H. Thymic output, T-cell diversity, and T-cell function in long-term human SCID chimeras Blood. 2009 114 7 1445 -1453
Speckmann C., Pannicke U., Wiech E., Schwarz K., Fisch P., Friedrich W., Niehues T., Gilmour K., Buiting K., Schlesier M., Eibel H., Rohr J., Superti-Furga A., Gross-Wieltsch U., and Ehl S. Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency Blood. 2008 112 10 4090 -4097
Speckmann C., Neumann C., Borte S., la Marca G., Sass JO., Wiech E., Fisch P., Schwarz K., Buchholz B., Schlesier M., Felgentreff K., Grimbacher B., Wiech E., Santisteban I., Bali P., Hershfield M.S., and Ehl S. J Allergy Clin Immunol. 2012 130 4 991 -994 Epub 2012 May 10
Villard J., Masternak K., Lisowska-Grospierre B., Fischer A., and Reith W. MHC class II deficiency: a disease of gene regulation Medicine (Baltimore). 2001 80 6 405 -418
Wolkewitz M., Allignol A., Schumacher M., and Beyersmann J. Two pitfalls in survival analyses of time-dependent exposure: a case study in a cohort of oscar nominees The American Statistician. 2010 64 3 205 -211
Wolkewitz M., Beyersmann J., Gastmeier P., and Schumacher M. Modeling the effect of time-dependent exposure on intensive care unit mortality Intensive Care Med. 2009 35 5 826 -832
Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G., Holland S.M., and Su H.C. Combined immunodeficiency associated with DOCK8 mutations N. Engl. J. Med. 2009 361 21 2046 -2055
Supplementary Material
Supplementary Data: Investigator Statement (lpsn-2015-0002suppl.pdf)
- Download
- 446.81 KB
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 2 • June 2015
Pages: 91 - 106
History
Received: 16 January 2015
Accepted: 28 January 2015
Accepted manuscript online: 17 February 2015
Version of record online: 17 February 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Carsten Speckmann MD, Annette Uhlmann PhD, Sam Doerken MSc, Martin Wolkewitz PhD, Annette Pohl, and Stephan Ehl MD. 2015. A prospective outcome study of patients with profound combined immunodeficiency (P-CID). LymphoSign Journal.
2(2): 91-106. https://doi.org/10.14785/lpsn-2015-0002
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
