Pulmonary hemorrhage in a case of CD25 deficiency
Abstract
Introduction: Presentation with severe autoimmune manifestations in early infancy is rare, especially when the gut is not involved.
Methods: Lymphocyte phenotyping, determination of autoantibodies, and Sanger sequencing were employed to investigate this case.
Results: A novel homozygous mutation in CD25 was identified in the patient who presented in the first weeks of life with insulin-dependent diabetes mellitus (IDDM) and subsequently developed autoimmune cytopenia and pulmonary hemorrhage.
Conclusion: CD25 deficiency is present in the Gulf (Oman) and cases with early autoimmune manifestations should be tested for this possibility.
Statement of novelty: A novel mutation in CD25 leads to an early presentation of IDDM and pulmonary hemorrhage.
Introduction
The interleukin-2 (IL-2) receptor consists of multiple subunits including the α chain (IL-2RA or CD25), the β chain (IL-2RB or CD122), and the γ chain (γc or IL-2RG or CD132). This receptor is critical for normal maturation, function, survival, and regulation of lymphocytes (Leonard et al. 1982; Sharon et al. 1986).
The most extensively studied γc chain binds IL-4, IL-7, IL-8, IL-15, and IL-21, whereas the β chain binds IL-15 and the α chain exclusively binds exclusively IL-2 (Teshigawara et al. 1987; O'Gorman et al. 2009).
IL2RA is expressed in induced or activated CD4+ and CD8+ T cells but is also constitutionally expressed at high levels in regulatory T cells (Tregs). Upon binding of IL-2, IL-2RA mediates and promotes the transcription of FOXP3 in a STAT5 dependent manner (Zorn et al. 2006).
The consequence of the loss of IL-2RA was first described by Sharfe et al. (1997) and Roifman (2000). IL-2RA deficiency resulted in combined immunodeficiency with susceptibility to viral and bacterial infections. Strikingly, patients suffered severe autoimmune manifestations including colitis, lymphadenopathy, hepatosplenomegaly, pneumonitis, and primary biliary cirrhosis (Aoki et al. 2006). The thymus in these patients was of normal size and populated with thymocytes, yet cortico-medullary structure was disrupted and Hassals corpuscles were absent (Sharfe et al. 1997). Apoptosis of thymocytes was dramatically reduced suggesting aberrant negative selection of thymocytes (Roifman, 2000). Subsequently, several genetic variants in CD25 were found to be associated with multiple autoimmune disorders (Lowe et al. 2007; Smyth et al. 2008; Alcina et al. 2009; Dendrou et al. 2009; Hinks et al. 2009).
This successful bone marrow transplant performed in this patient demonstrated for the first time that the variety of autoimmune features can be completely reversed by allogeneic hemopoetic stem cell transplant.
Subsequently, an 8-year-old patient with colitis, pneumonitis, and insulin-dependent diabetes mellitus (IDDM) was found to have mutations in CD25. It was demonstrated in this case that IL-2RA was absolutely required for IL-10 secretion.
Another 8-year-old patient with IL-2RA deficiency was recently reported (Caudy et al. 2007). She had bullous pemphigoid at 1 year of age, autoimmune thyroiditis at 4 years of age, and she developed severe colitis with Cytomegalovirus (CMV) infections at 5 years of age (Goudy et al. 2013).
We report here for the first time a patient from Oman with a novel mutation in CD25. The presentation was both very early in life and severe in nature.
Methods
Flow cytometry
Peripheral blood cells were stained with monoclonal anti-CD25 conjugated with phycoerythrin or with isotype-matched control antibodies. They were analyzed on a Becton Dickenson flow cytometer.
Sequence analysis
Genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by polymerase chain reaction (PCR) with specific primers for exons 1–8 of the coding sequence of IL2RA (NM_000417.2) (available upon request). Exons and their flanking intronic regions were sequenced with the GenomeLab Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter), and analyzed on a CEQ 8000 Genetic Analysis System (Beckman Coulter).
Results
Case report
A male infant patient, born to consanguineous parents after a normal pregnancy and delivery, was diagnosed with IDDM as well as hypothyroidism. Antibodies to glutamic acid decarboxylase, anti-islet cells, and antiperioxidase were positive in his blood. He was therefore treated with replacement insulin and thyroxine.
He was admitted to a community hospital at 10 months of age and presented with a 3-month history of intermittent fevers as well as abdominal distension due to massive enlargement of the liver and spleen. Laboratory investigation identified a urinary tract infection with Enterococcus faecalis as well as a positive detection of CMV DNA by PCR. He was treated with antibiotics and antiviral agents with no improvement. He was subsequently referred to a tertiary centre that identified autoimmune liver disease with increased conjugated bilirubin and positive autoantibodies to smooth muscle and to liver/kidney microsome. He constantly suffered Coombs-positive hemolytic anemia and thrombocytopenia with intermittent neutropenia. Examination of bone marrow aspirate revealed a normal representation of all hematopoietic lineages.
He subsequently deteriorated, developing respiratory failure and requiring assisted ventilation. Chest X-rays showed bilateral interstitial infiltrates, and bronchoscopy revealed profuse fresh blood arising from multiple lung lobes. Bronchoalveolar lavage tested for multiple pathogens was negative for bacteria, Pneumocystis jiroveci, and multiple viruses. Because the patient had high titres of cytoplasmic anti-neutrophil cytoplasmic antibodies (c-ANCA; 184 Ru/mL), perinuclear-ANCA (199 Ru/mL), and anti-glomerular basement membrane antibodies, it was assumed that the he suffered small vessel pulmonary vasculitis that contributed to the lung bleeding.
He was treated with a pulse of methylprednisolone as well as daily steroids, four doses of rituximab, and one dose of cyclophosphomide that was discontinued because of profound neutropenia and septicemia. In addition, he was subsequently treated with mycophenolate mofetil (MMF) demonstrating gradual improvement of lung disease, cessation of bleeding, and was weaned off ventilation. Additionally, the patient's blood counts and bilirubin levels normalized following this treatment. He remains on daily MMF and 1 mg/kg prednisone and is currently awaiting hematopoietic stem-cell therapy.
Evaluation of the immune system
Flow cytometry analysis at presentation was normal. However, expression of IL-2RA was undetectable on CD4+ or CD3+ cells (Figure 1).
Figure 1:
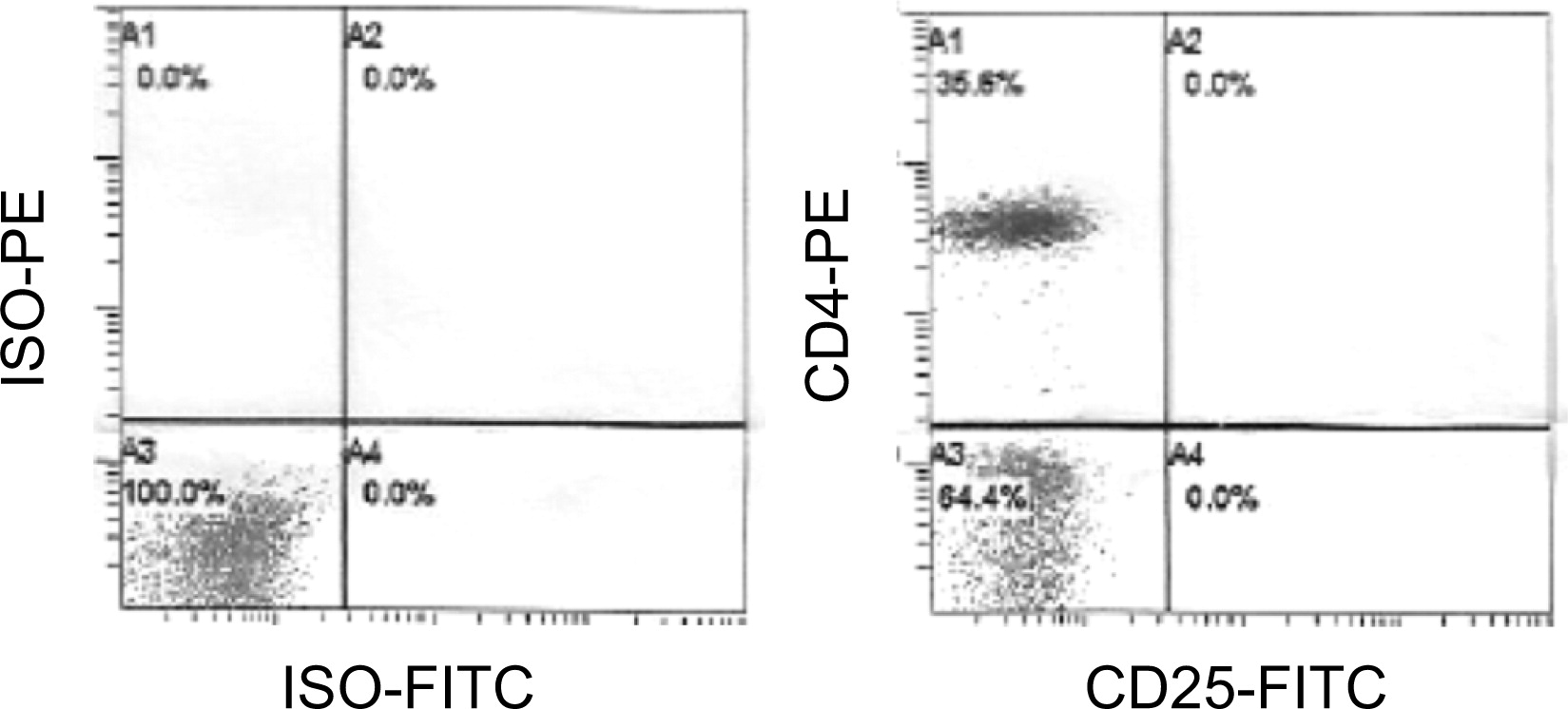
The immunoglobulin levels were normal for the patient's age.
Analysis of the CD25 gene
Sanger sequencing of CD25 revealed a homozygous T to C substitution at position 418 c.418 T > C, which would predict a tyrosine to histidine change in the extracellular portion of IL-2RA. This amino acid substitution is located in the SUSHI-2 domain or in a complement control protein module. This mutation clearly affected the stability of the protein, as no IL-2RA protein was detected upon CD3+ or CD4+ cells (Figure 2).
Figure 2:
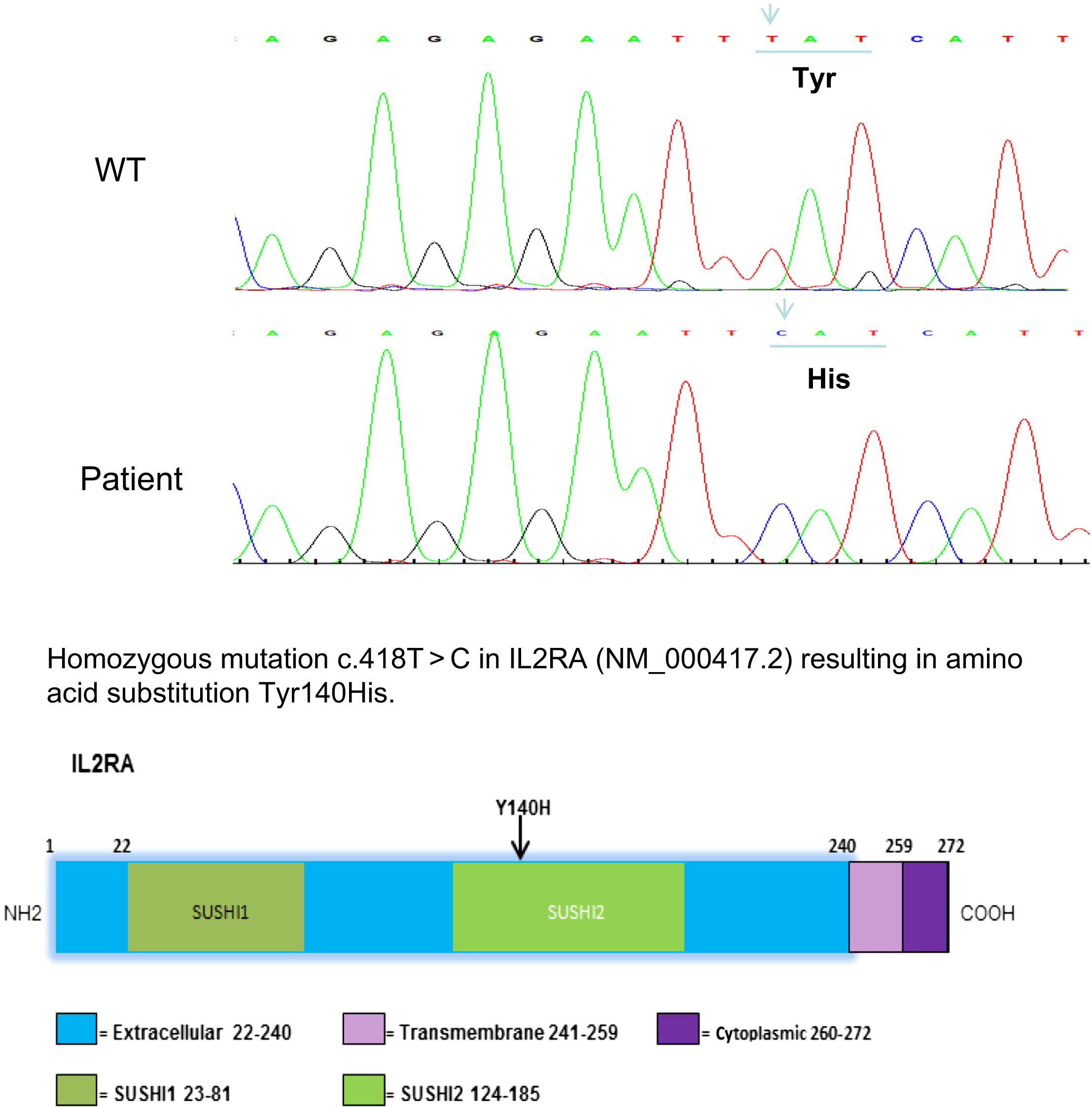
Discussion
To our knowledge this is the first case report about a patient with CD25 deficiency born to parents from Oman. The homozygous c. 418 T > C mutation is novel and indicates a founder effect in this region.
Of the patients known to have CD25 deficiency, whether reported or not (personal communications), this patient presented very early in life, almost immediately after birth with severe autoimmune manifestations including IDDM as well as autoimmune hemolytic anemia, neutropenia, and thrombocytopenia.
Uniquely, the patient in this case developed life threatening pulmonary hemorrhage and unlike all cases known to date had no evidence of colitis.
However, this case highlights the features common to all cases of CD25 deficiency. Invariably, all cases have CMV disease involving the lungs with or without infection of the gut. Commonly, lymphocytic infiltrates are detected in multiple organs. Lymphadenopathy and hepatosplenomegaly were reported in all cases, whereas thyroid disease and eczema were identified in some but not all cases.
The presentation with infantile colitis may raise the possibility of other monogenic disorders such as deficiencies of IL-10RA, IL-10RB, IL-10, or forkhead box P3 (FOXP3). However unlike CD25 deficiency, the lack of universal deficiency in T cells (normal T-cell receptor excision circles (TRECS), normal thymus) and susceptibility to infections is common to these disorders. Conditions that can combine profound T-cell deficiency and autoimmune manifestations including enteropathy and endocrine abnormalities are TTC7A and STAT1 deficiencies.
A feature that distinguishes CD25 deficiency from all these disorders is the heavy infiltrates of lymphocytes in target organs, probably triggering tissue damage. As described in the first case (Sharfe et al. 1997) infiltrates were detected in the lungs, gut, and the liver. This feature suggests that infiltrating T cells are autoreactive in nature. There are several mechanisms involved in creating and propagating such cells in CD25 deficiency. The first is the abnormality in central tolerance. The thymus in CD25 deficiency showed reduced expression of CD1 that lead to abnormal negative selection coupled with decreased apoptosis that resulted in prolonged survival of these autoreactive T cells (Roifman 2000).
The second mechanism is the impaired peripheral regulation of T cells because of reduced CD25+ FOXP3+ CD4+ Tregs. Unlike FOXP3, IL10RA, IL10RB, and IL10 deficiencies, a lack of CD25 causes combined immunodeficiency as well as autoimmunity. Indeed, all known cases suffer severe CMV infections of the lung and gut.
In summary, we reported a unique case of CD25 deficiency caused by a novel mutation. Presentation was very early in infancy and included pulmonary hemorrhage but without symptoms of colitis.
REFERENCES
Alcina A., Fedetz M., Ndagire D., Fernandez O., Leyva L., Guerrero M., et al. IL2RA/CD25 gene polymorphisms: uneven association with multiple sclerosis (MS) and type 1 diabetes (T1D) PLoS One. 2009 4 e4137
Aoki C., Roifman C.M., Zhe-Xiong L., Bowlus C.L., Norman G.L., Shoenfeld Y., et al. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis Journal of Autoimmunity. 2006 27 50 -53
Caudy A.A., Reddy S.T., Chatila T., Atkinson J.P., and Verbsky J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes J. Allergy Clin. Immunol. 2007 119 482 -487
Dendrou C.A., Plagnol V., Fung E., Yang J.H., Downes K., Cooper J.D., et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource Nat. Genet. 2009 41 1011 -1015
Goudy K., Aydin D., Barzaghi F., Gambineri E., Vignoli M., Mannurita S.C., et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity Clin. Immunol. 2013 146 248 -261
Hinks A., Ke X., Barton A., Eyre S., Bowes J., Worthington J., et al. Association of the IL2RA/CD25 gene with juvenile idiopathic arthritis Arthritis Rheum. 2009 60 251 -257
Leonard W.J., Depper J.M., Uchiyama T., Smith K.A., Waldmann T.A., and Greene W.C. A monoclonal antibody that appears to recognize the receptor for human T-cell growth factor; partial characterization of the receptor Nature. 1982 300 267 -269
Lowe C.E., Cooper J.D., Brusko T., Walker N.M., Smyth D.J., Bailey R., et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes Nat. Genet. 2007 39 1074 -1082
O'Gorman W.E., Dooms H., Thorne S.H., Kuswanto W.F., Simonds E.F., Krutzik P.O., et al. The initial phase of an immune response functions to activate regulatory T cells J. Immunol. 2009 183 332 -339
Roifman C.M. Human IL-2 receptor α chain deficiency Pediat. Res. 2000 48 6 -11
Sharfe N., Dadi H.K., Shahar M., and Roifman C.M. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor Proc. Natl. Acad. Sci U.S.A. 1997 94 3168 -3171
Sharon M., Klausner R.D., Cullen B.R., Chizzonite R., and Leonard W.J. Novel interleukin-2 receptor subunit detected cross-linking under high-affinity conditions Science. 1986 234 859 -863
Smyth D.J., Plagnol V., Walker N.M., Cooper J.D., Downes K., Yang J.H., et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease N. Engl. J. Med. 2008 359 2767 -2777
Teshigawara K., Wang H.M., Kato K., and Smith K.A. Interleukin 2 high-affinity receptor expression requires two distinct binding proteins J. Exp. Med. 1987 165 223 -238
Zorn E., Nelson E.A., Mohseni M., Porcheray F., Kim H., Litsa D., et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo Blood. 2006 108 1571 -1579
Information & Authors
Information
Published In

LymphoSign Journal
Volume 01 • Number 01 • September 2014
Pages: 39 - 43
History
Received: 22 May 2014
Accepted: 22 May 2014
Accepted manuscript online: 2 July 2014
Version of record online: 2 July 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Al SukaitiNashat, Al SinaniAisha, Al IsmailySuad, ShaikhSamiuddin, and Al AbrawiSafia. 2014. Pulmonary hemorrhage in a case of CD25 deficiency. LymphoSign Journal.
01(01): 39-43. https://doi.org/10.14785/lpsn-2014-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
