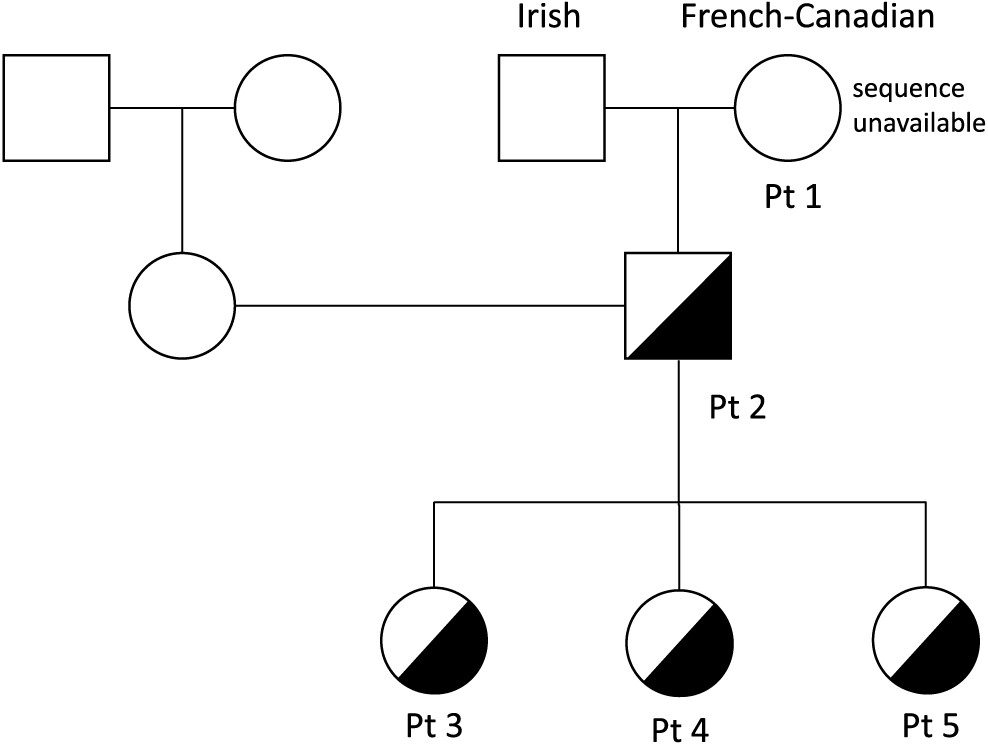
Patient 1
Patient 1 was a product of non-consanguineous parents of Irish and French descent. She presented at young adulthood, age 23 years, with recurrent prolonged upper respiratory tract infections (10/year), chest X-ray-proven pneumonias (5/year), chronic sinusitis, and recurrent otitis with purulent otorrhea, fatigue, and weight loss. Her childhood clinical history was unremarkable, with uneventful chickenpox, measles, and mumps infections. She had 1 episode of arthritis at age 2 years and normal echocardiography on examination. A tonsillectomy was performed at 5 years of age.
During her initial evaluation with our Immunology team at age 26 years, she presented with cylindrical bronchiectasis on bronchoscopy of the left lower lobe and the lingula. She had normal tuberculin test results and sweat chloride levels (7 mEq/L). Immunological evaluation (
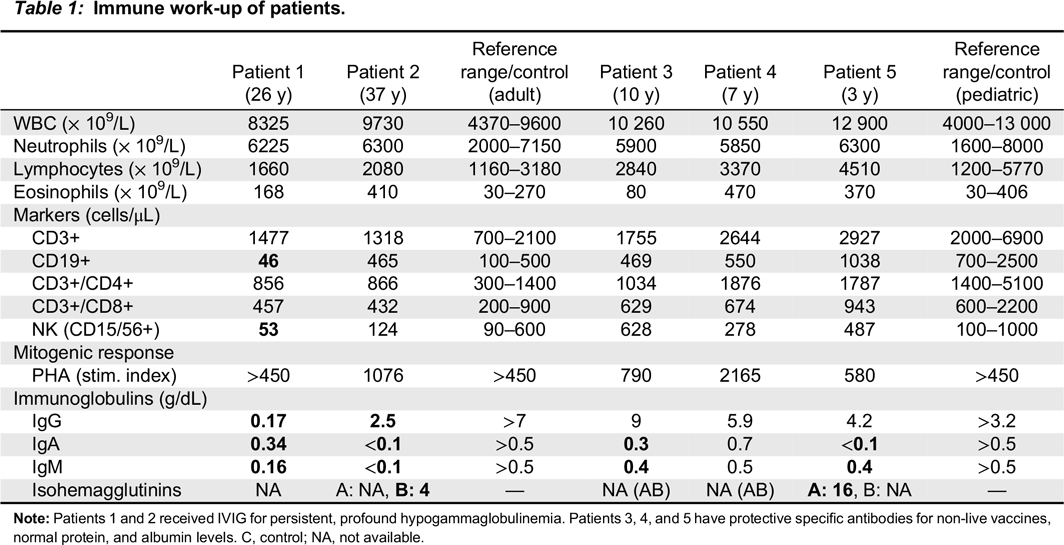
Table 1) showed pan-hypogammaglobulinemia, with low IgG, IgM, and IgA levels, as well as low anti-tetanus toxoid titer (0.03 ng/mL). IgE levels were normal, as were protein and albumin levels. Her CBC and differential was unremarkable. Lymphocyte counts were normal, however, B cell counts were almost absent with only 46 cells/μL of CD19+ cells alongside decreased NK cell numbers of 53 cells/μL. Assay for spontaneous suppressive cell activity assay was positive, and E-rosette and AET rosette assessment of T lymphocytes were normal at 58% and 70%, respectively. Results of mixed lymphocyte reaction were also normal. Lymphocyte proliferation responses to all mitogens except STA were normal, however, antigen stimulation responses to
Candida and Tuberculin PPD were decreased.
Due to her clinical presentation and diminished immune responses, she was started on fresh frozen plasma (FFP), the only IgG replacement option that was available at the time she was diagnosed. She was eventually transferred to intramuscular and then intravenous immunoglobulin therapy. Laboratory assessments showed adequate IgG replacement. Unfortunately, the patient contracted blood product-related Hepatitis C infection resulting in liver cirrhosis, portal hypertension, massive ascites and varicose veins requiring multiply ligation and sclerotherapy due to bleeding, and eventually liver failure. Other immunodeficiency related clinical manifestations include chronic cough, yellow sputum, Mycoplasma colonization, chronic Salmonella gastroenteritis which resolved after 6 months of antibiotic therapy, chronic non-infectious diarrhea, and hepatosplenomegaly.
At the age of 36 years, the patient developed ophthalmic melanoma resulting in loss of vision in the affected eye. She suffered severe bronchiectasis with pyocele 2 years later for which she underwent left lower lobe lobectomy. Concurrently, she slowly developed progressing cerebellar symptoms with abnormal Romberg and heel/toe gait, however, CT imaging of the head was unremarkable.
Following a prolonged course of weight loss, intermittent fevers, and 1 episode of septicaemia, worsening productive cough, failed empiric antibiotic therapy, and increased fatigue, the patient was subsequently evaluated for malignancy. She was diagnosed with lymphoma and later died at age 45 years.
Patient 2
Patient 2, the son of patient 1, was evaluated at 1 year of age and found to have low IgG, IgM, and IgA levels (
Table 1). At the age of 10 years he developed chronic immune thrombocytopenia and has been on regular monthly intravenous IgG replacement therapy since age 11 years. He was diagnosed with persistent abdominal lymphadenomegaly, and percutaneous needle aspiration biopsy showed follicular hyperplasia. His CBC and differential were unremarkable, with normal numbers of circulating CD3+, CD4+, CD8+, CD19+ and NK cells. Stimulation of T cells with anti-CD3 and anti-CD28 was markedly reduced, implying a T cell defect. In vitro lymphocyte responses to a series of antigens, including
Candida species, tetanus toxoid, zoster, and cytomegalovirus, were also extremely low indicating the antigen specific memory T cells were either lacking or present at low frequencies (
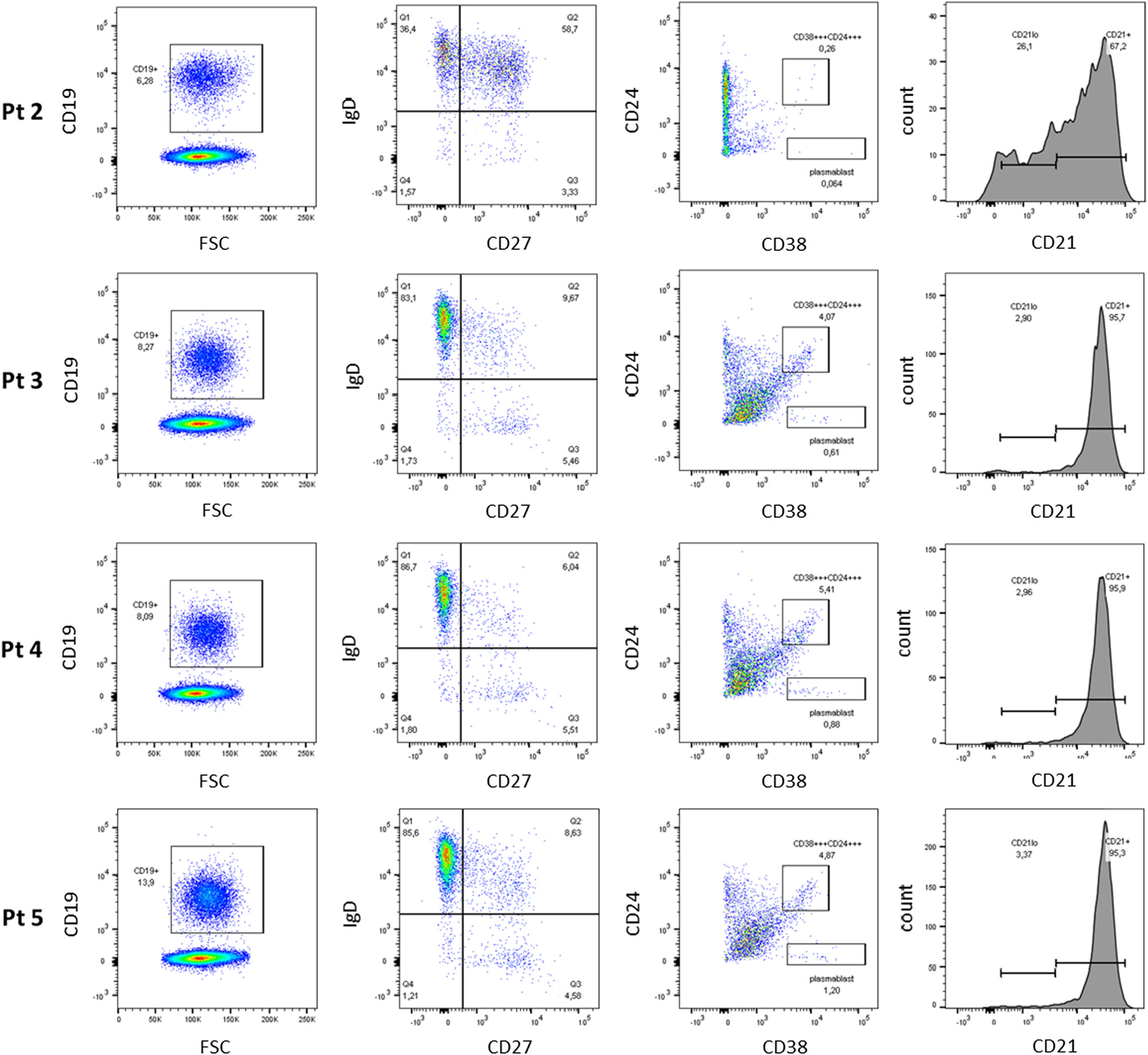
Figure 2). Assessment of B cell phenotype by flow cytometry showed increased number of IgD+ memory cells, but diminished class switching (CD38+/CD24+) (
Figure 3). No transitional B cells and elevated number in CD21
low B cells were detected.
Patient 3
Patient 3, daughter of patient 2, is a 10 year old with a history of 3 episodes of chickenpox at ages 3, 5, and 7 years. Given the clinical history of her grandmother and father, live viral vaccines were avoided. Her CBC and differential were unremarkable, with normal B and T cell lymphocyte function. She had normal IgG levels with adequate specific antibody titers, but decreased levels of IgA and IgM. Her varicella zoster virus specific IgG antibody titers were negative. Despite 3 episodes of chickenpox, she was unable to mount an appropriate immune response.