A novel mutation in LIG4 in an infant presenting with severe combined immunodeficiency with thymic medullary dysplasia
Abstract
Background: DNA ligase IV deficiency is a rare autosomal recessive condition resulting from mutations in LIG4, an essential component of the non-homologous end-joining pathway that prevents mutagenesis and apoptosis. Patients with LIG4 deficiency present with varying degrees of combined immunodeficiency, or less commonly, severe combined immunodeficiency (SCID). Assessment of thymus pathology has been instrumental in defining a growing number of T cell deficiencies. In this case report, we present thymic histopathology of a LIG4 deficient patient who presented with SCID.
Methods: Whole exome sequencing and Sanger confirmation were used to identify a novel mutation in LIG4. Standard immune work up and histopathology were completed to characterize deficits in immune function and dysplastic thymic architecture in our patient.
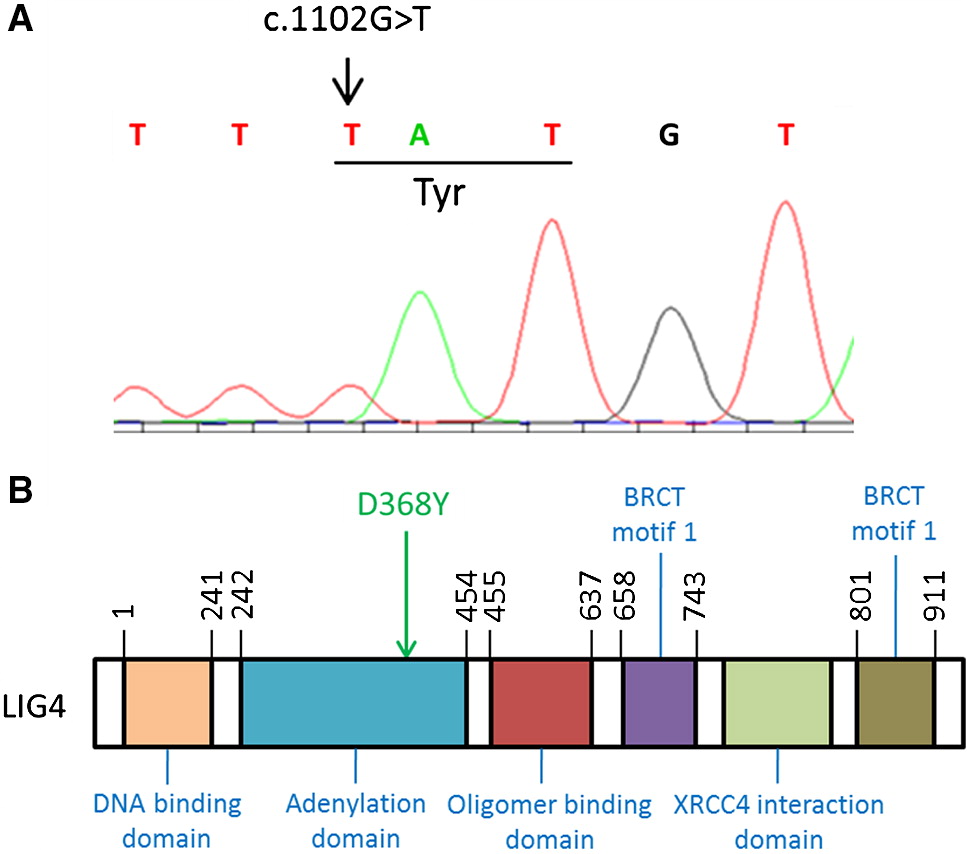
Results: Next generation sequencing techniques identified a homozygous c.1102G>T, resulting in amino acid change D368Y in the adenylation domain of LIG4. Histopathology revealed a distinct absence of Hassall’s corpuscles, lack of cortico-medullary demarcation, as well as lack of T cells and Langerhans histiocytes in the thymic medulla.
Conclusion: We have identified a novel mutation in LIG4 resulting in a SCID phenotype. Underdevelopment of the thymus, characterized by a lack of Hassall’s corpuscles and competent thymocytes, likely contributes to the immune defects observed in patients with mutations in LIG4.
Statement of novelty: We report here a novel mutation in LIG4 as well as the first description of detailed thymus pathology in this condition.
Introduction
DNA ligase IV (LIG4) has an essential role in T and B cell development. LIG4 is involved in the final step of the ubiquitous non-homologous end-joining (NHEJ) repair pathway involved in V(D)J recombination, which contributes to the extensive diversity seen in the formation of T cell receptors (TCRs) and immunoglobulins.
V(D)J recombination is initiated with the creation of double stranded breaks (DSBs) at the ends of the variable (V), diversity (D), and joining (J) segments of TCR and immunoglobulin genes. This process is achieved with the binding of recombination-activation gene 1 (RAG1) and RAG2 to recombination signal sequences. The ends of the DSBs are first stabilized by proteins Ku70 and Ku80. Following this, DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is recruited to the complex and activates Artemis, which opens up the hairpin loops at the DSBs, allowing the random addition and deletion of nucleotides by the enzyme terminal deoxynucleotidyl transferase (Tdt). This further increases the genetic diversity of TCRs and immunoglobulins. Finally, the ends are ligated together by LIG4 in association with its cofactors XRCC4 and Cernunnos/XLF (de Miranda et al. 2011).
The defect of any of these proteins may lead to aberrant V(J)D recombination and varying degrees of immunodeficiency. The NHEJ repair pathway is also essential in repairing damaged DNA, induced by ionizing radiation. For this reason, abnormal expression of proteins involved in NHEJ repair often predispose affected individuals to cellular radiosensitivity as well as malignancy (de Miranda et al. 2011).
Mutations in LIG4 cause a rare autosomal recessive condition termed LIG4 deficiency (OMIM 606593). The first patient with a mutation in LIG4 was reported in 1990 by Plowman and colleagues (Plowman et al. 1990). Patients present with varying degrees of combined immunodeficiency, microcephaly, growth failure, developmental delay, pancytopenia, and malignancy. The degree of immunodeficiency ranges from clinically asymptomatic to severe combined immunodeficiency (SCID). Among the reported cases, only 6 presented with a SCID phenotype (Buck et al. 2006; Enders et al. 2006; van der Burg et al. 2006; Grunebaum et al. 2008).
The interaction between developing T cells and thymic epithelial cells is essential for the formation of thymic architecture, and comprises 2 components, the cortical thymic epithelial cells and the medullary thymic epithelial cells. The medullary thymic epithelial cells give rise to the development of the Hassall’s corpuscles. This developmental pathway is mediated via lymphotoxin signals from positively selected thymocytes, as well as other mediators, such as AIRE (autoimmunume regulator). AIRE contains an enhancer element that acts as a molecular switch to promote the differentiation of medullary thymic epithelial cells (Yano et al. 2008).
Mouse models of RAG1 and RAG2 have revealed significant structural abnormalities in the thymus, including the loss of the cortico-medullary demarcation (Rucci et al. 2011; Marrella et al. 2014). In contrast, a mouse model for LIG4 deficiency (R278H) found preserved cortico-medullary demarcation (Rucci et al. 2011). Unfortunately, the thymus structure in mice remains a poor model for human thymic morphology, mostly because of the lack of formed Hassall’s corpuscles. The presence of fully formed Hassall’s corpuscles is the hallmark for normal human thymus architecture. So far, there have been no reports describing a lack of Hassall’s corpuscles in an immunologically competent normal individual. Furthermore and importantly, lack of or poorly developed Hassall’s corpuscles have been shown to be associated with profound immunodeficiencies, such as SCID (Roifman 2005). Various degrees of thymus dysplasia have been previously reported in multiple SCID types, including Zap-70 deficiency (Roifman et al. 1989), CD35 deficiency (Dadi et al. 2003), IL-2Rα (Sharfe et al. 1997), RelB deficiency (Sharfe et al. 2015), RMRP cartilage hair hypoplasia (Roifman et al. 2006), and others (Ratech et al. 1985; Poliani et al. 2009).
To date, thymus histopathology has not yet been described in LIG4 deficiency. Here, we report a case of LIG4 deficiency in an infant who presented with SCID almost 20 years ago. Using the technology of whole exome sequencing, we identified a novel mutation in LIG4. We also describe, for the first time, the histological features of the thymus in a patient with LIG4 deficiency.
Methods
Lymphocyte markers and T cell proliferative responses
Blood mononuclear cells obtained by Ficoll-Hypaque density gradient centrifugation were assessed for surface phenotypes using direct immunofluorescence. Fluorescein isothiocyanate-conjugated goat anti-human immunoglobulin antibody (Tago, Burlingame, CA) or fluorescein isothiocyanate-conjugated monoclonal antibodies to anti-CD3, CD4, CD8, CD20, and CD56 (Coulter Instruments, Mississauga, Ontario, Canada) were utilized. Analysis was performed on a Coulter EPICS V flow cytometer. Lymphocyte proliferative responses to mitogens, including phytohemagglutinin (PHA), Staphylococcus aureus (STA), and Staphylococcal-protein A (SpA), were determined by 3H-Thymidine incorporation. All assays were performed in triplicate and compared with normal controls (Arpaia et al. 1994).
Serum concentration of immunoglobulins
Serum concentrations of immunoglobulins were measured by nephelometry, as previously described (Atkinson and Roifman 2007).
Whole exome sequencing and variant calling
DNA from blood was submitted to The Centre for Applied Genomics (TCAG), Toronto, Canada for exome library preparation and sequencing. DNA was quantified by Qubit DNA HS assay (Life Technologies, Carlsbad, CA) and 100 ng of input DNA was used for library preparation using the Ion AmpliSeq Exome Kit (Life Technologies) according to the manufacturer’s recommendations. The Ampliseq Exome library was immobilized on Ion PI™ Ion Sphere™ particles using the Ion PI Template OT2 200 Kit v3. Sequencing was performed with the Ion PI Sequencing 200 Kit v3 and Ion PI Chip v2 in the Ion Proton™ semiconductor sequencing system following the manufacturer’s recommendation.
Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server, using the Ion Proton ampliseq germline low stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline (Stavropoulos et al. 2016) based on Annovar (November 2014 version) (Wang et al. 2010) and RefSeq gene models (downloaded from UCSC 1 August 2015).
Variant prioritization
High quality variants were prioritized (Bamshad et al. 2011; Merico 2016) based on (i) allele frequency based on the public databases 1000 Genomes (Auton et al. 2015), ExAC (Lek et al. 2016), Wellderly (Erikson et al. 2016) and in-house Ion Proton™ AmpliSeq platform-matched unaffected controls (Tammimies et al. 2015); (ii) clinical classification as pathogenic or likely pathogenic, if available in the public databases ClinVar (Landrum et al. 2016) and HGMD (Stenson et al. 2014); (iii) genomic conservation based on the phyloP (Pollard et al. 2010) and PhastCons (Siepel et al. 2005) scores, effect and predicted damaging impact on the gene product based on the predictors SIFT (Ng and Henikoff 2001), PolyPhen2 (Adzhubei et al. 2010), MutationAssessor (Reva et al. 2011), CADD (Kircher et al. 2014), Spidex (Xiong et al. 2015); and (iv) gene implication in human immune disorder and Mendelian mode of inheritance based on the Clinical Genomics Database (CGD) (Solomon et al. 2013), the Human Phenotype Ontology (HPO) (Kohler et al. 2014), Online Mendelian Inheritance in Man (OMIM; https://omim.org/), abnormal immune or hematopoietic phenotype phenotype in mouse based on the Mammalian Phenotype Ontology (MPO) annotations (Smith and Eppig 2012) provided by the Mouse Genome Informatics (MGI) database (Eppig et al. 2015), immune gene function annotations based on Gene Ontology (Blake et al. 2013) and pathway databases (Fabregat et al. 2016; Kanehisa et al. 2016), and constraint to truncating or missense mutation for heterozygous variants provided by ExAC (Lek et al. 2016). Minimum damaging impact for missense variants was defined as at least 2/6 impact predictors and phyloP conservation passing threshold for damaging impact (Stavropoulos et al. 2016). OMIM morbidmap annotations were downloaded in January 2015; Gene Ontology and pathway annotations were downloaded in March 2013; MPO/MGI and HPO annotations were downloaded in June 2015.
Pathology
Thymus removed at the time of autopsy was fixed in 10% buffered formalin for 24 hours. The tissue was dehydrated and then processed in organic solvents for embedding in paraffin. 3 to 5 micron sections of the tissue were prepared for hematoxylin and eosin staining. For immunohistochemistry staining, monoclonal antibodies (FLEX-ready to use system of monoclonal antibodies purchased from Dako, CA) were utilized, following immunostaining conditions and procedures recommended for use with the automated immunohistochemistry staining equipment (DAKO Omnis, Agilent, Mississauga, Ontario, Canada). Appropriate normal tissue controls were used to verify the accuracy of the immunostaining results.
Results
Case report
Our patient is a female infant born in Ontario in 1998 to consanguineous parents of Pakistani descent. She was born at 40 weeks gestation, weighing 2.8 kg (15th percentile), following an uncomplicated pregnancy and delivery. She presented to our institution at 4 months of age with an acute multilobar pneumonia. She also had a history of severe eczema since 6 weeks of age, failure to thrive with weight dropping to below the 3rd percentile for age, and microcephaly (head circumference 36.5 cm, below the 3rd percentile for age). Initial nasopharyngeal swab grew Parainfluenza virus type III. During the course of admission, bronchoalveolar lavage also isolated Pseudomonas aeruginosa and Pneumocystis jirovecii, and blood culture was positive for Pseudomonas aeruginosa. Immune evaluation was consistent with T-B-NK+ SCID. Human leukocyte antigen (HLA) typing was performed but the donor search was unable to identify a suitable matched sibling or matched unrelated donor. Due to the ongoing deterioration of our patient’s respiratory status, we proceeded with peripheral blood stem cell transplantation from the patient’s mother, who was a 4/6 haploidentical donor (A and B mismatch) with CD34+ selection. She underwent pre-transplant conditioning consisting of busulfan and cyclophosphamide. Unfortunately, our patient further deteriorated during conditioning and died from respiratory failure just prior to transplant at 8 months of age.
Genetic evaluation
Utilizing whole exome sequencing and Sanger confirmation, we identified a novel homozygous mutation c.1102G>T in LIG4 (NM_206937), resulting in a D368Y amino acid substitution (Figure 1A). The novel variant described had a frequency of 0 in control databases, missense damaging tier-2 impact, and occurred in a gene implicated in human disorder (LIG4; locus:chr13:108862515:108862515:C:A). This mutation is in the adenylation domain of LIG4 (Figure 1B) and has not previously been reported in the literature.
Figure 1:
Immune evaluation
Immune work up was significant for lymphocytopenia (0.46 × 109 cells/L) with lymphocyte immunophenotyping showing near-absent CD19+ cells (1 cell/µL), reduced CD4+ cells (148 cells/µL) and reduced CD8+ cells (30 cells/µL). CD56+CD16+ cells were within normal limits at 93 cells/µL. Immunoglobulins were low, with IgG 0.5 g/L, IgA <0.1 g/L, and IgM 0.3 g/L. Mitogen stimulation showed no response to PHA, STA, and SpA.
Thymus pathology
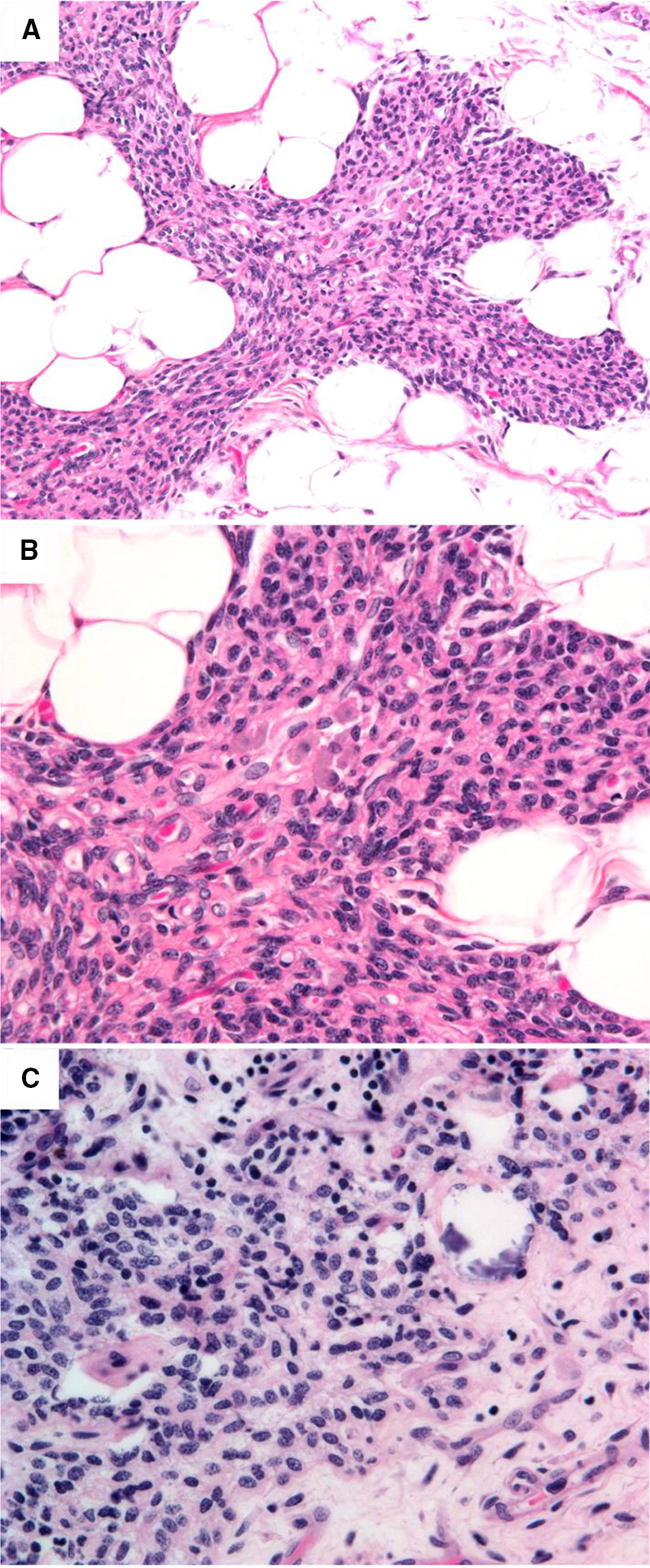
Thymus histopathology from the patient’s autopsy revealed a significantly underdeveloped thymus with increased fat infiltration. The patient’s thymus was compared with autopsy sections of thymus from an immunocompetent patient, who had also received a chemotherapeutic regimen in preparation for bone marrow transplantation. Histopathology of the thymus from both our patient with LIG4 deficiency, as well as the control, showed near complete absence of thymic T and B cells, but not macrophages. As a result, there was marked shrinkage of the thymus cortical epithelial reticulum. In the LIG4 deficient patient, the most significant observation was the presence of medullary thymic epithelial dysplasia, leading to the loss of cortico-medullary demarcation and the complete absence of Hassall’s corpuscles (Figures 2A, 2B). By comparison, a compact collapsed thymic epithelial reticulated network and Hassall’s corpuscles can be seen in the medulla of the control thymus (Figure 2C).
Figure 2:
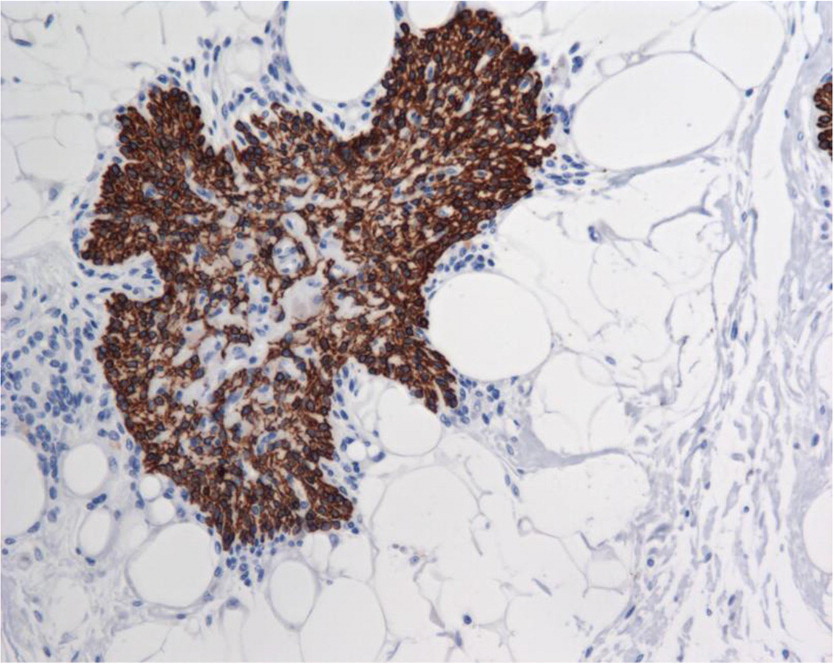
Immunohistochemistry for cytokeratin identified the presence of both cortical and medullary thymic epithelial cells, which form a mesh-like reticulum network. However, development of Hassall’s corpuscles, as evidenced by concentric whorls of hyperplasic cytokeratin-positive hyperplastic medullary thymic epithelial cells, was absent (Figure 3).
Figure 3:
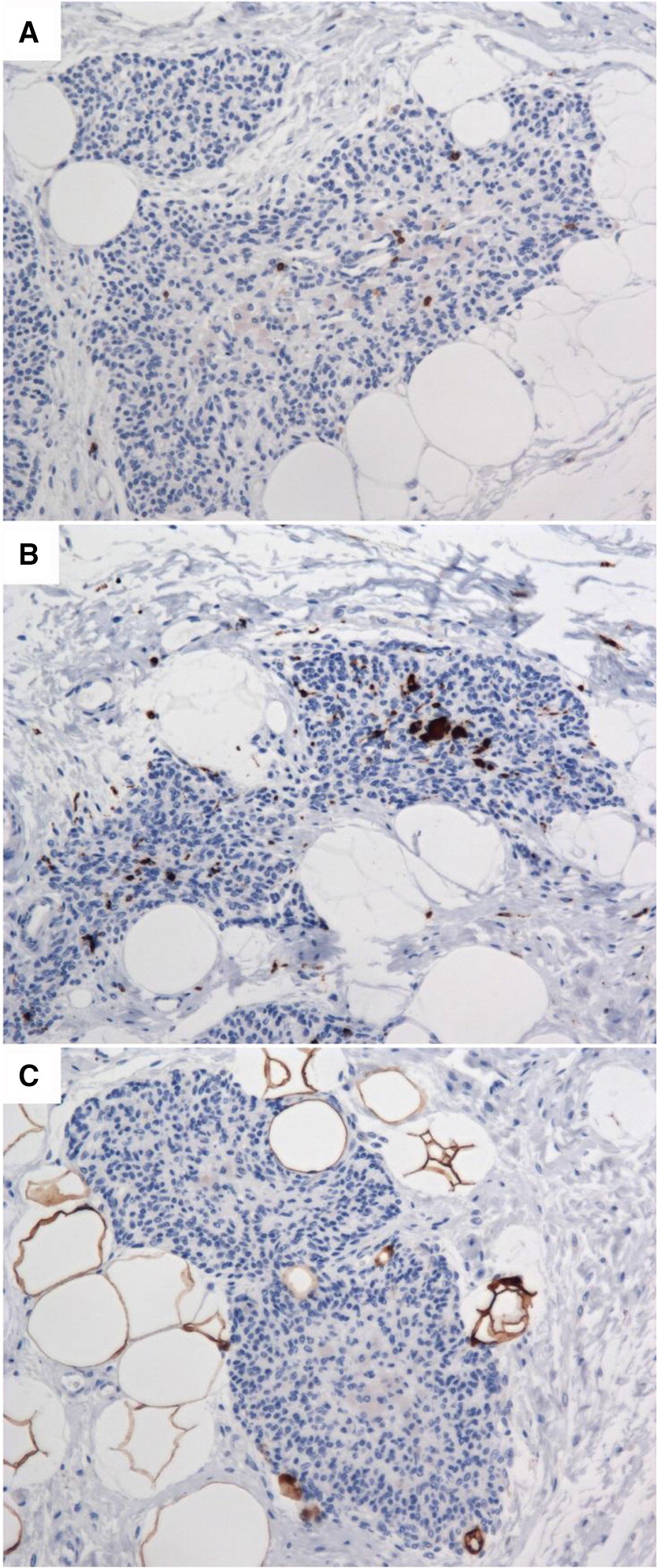
Immunostaining for T lymphocytes using an anti-CD3 antibody showed near total depletion of lymphocytes (Figure 4A). CD4 and CD8 staining was also absent (not shown); although this is not unexpected in the context of post-conditioning with chemotherapy. As seen in the hematoxylin and eosin stained thymus sections (Figure 2B), and verified by immunostaining for CD68 (Figure 4B), the thymic medulla contains only macrophages. There was also an absence of Langerhans histiocytes (negative S100 staining results, as seen in Figure 4C).
Figure 4:
Discussion
Damage to DNA integrity is part of normal homeostasis or cellular changes involved in meiosis, cell division or immunoglobulin and T cell receptor recombination (Yang et al. 2016). Increased damage may also be inflicted by ionizing radiation or chemical compounds. To maintain genomic integrity, a safety net of DNA repair pathways has evolved. In lymphocytes, a major mechanism of DNA repair is non-homologous end-joining, which is required for the DNA double strand break repair. The NHEJ comprises a complex of 5 proteins including DNA-PKcs, Ku70, Ku80 as well as XRCC4 and LIG4 (Yang et al. 2016). Impairment in any of these proteins results in ablated capacity to carry out V(D)J gene rearrangement that is essential for creation of a full TCR and BCR repertoire (Slatter and Gennery 2010).
Deleterious mutations in DNA repair components result in pleiotropic phenotypes including immunodeficiency, susceptibility to cancer, neurodegeneration and developmental abnormalities (de Miranda et al. 2011).
LIG4 deficiency is a rare autosomal recessive syndrome comprising of combined immunodeficiency, microcephaly, developmental delay and predisposition to cancer (Plowman et al. 1990; Hayani et al. 1994; Riballo et al. 1999; O’Driscoll et al. 2001; Buck et al. 2006; Enders et al. 2006; van der Burg et al. 2006; Gruhn et al. 2007; Toita et al. 2007; Grunebaum et al. 2008; Unal et al. 2009; IJspeert et al. 2013; Yue et al. 2013; Murray et al. 2014). Less than 50 patients have been described so far with the majority having various degrees of T cell deficiency and hypogammaglobulinemia. Furthermore, only a few have been reported to present early in infancy with features of SCID (Buck et al. 2006; Enders et al. 2006; van der Burg et al. 2006) or Omenn Syndrome (Grunebaum et al. 2008).
Strict phenotype/genotype correlation has not been established, but most severe SCID cases were caused by truncating mutations resulting in a shorter protein, rather than point mutations associated with CID or no significant immunodeficiency.
We describe here a patient with typical features of SCID who was prepared for bone marrow transplantation before the molecular diagnosis of LIG4 deficiency was available. Two decades later, WES was employed to reveal the genetic aberration and identified a homozygous novel mutation in the adenylation domain. Interestingly, this domain appears to pathogenic mutation hotspot, with another 3 pathogenic/likely pathogenic missense mutations and 1 non-frameshift single amino acid deletion found on ClinVar.
An excellent prediction of the severity of T cell deficiency is the presence of a dysplastic thymus. Typically, the morphology of a SCID thymus consists of a complete disruption of the thymic architecture, including loss of cortico-medullary structure as well as absence of Hassall’s corpuscles. This case provided a unique opportunity to compare the structure of the thymus in a primary as opposed to secondary immunodeficiency (chemotherapy for cancer). While chemotherapy in a cancer case depleted thymocytes, it didn’t markedly affect the presence of Hassall’s corpuscles. These results attest to the notion that a lack of Hassall’s corpuscles is highly predictive of a profound T cell immunodeficiency.
In summary, we have described here a patient with SCID due to a novel point mutation in LIG4, and characterized for the first time, the pathology of the thymus in this condition.
REFERENCES
Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., and Sunyaev S.R.2010. A method and server for predicting damaging missense mutations. Nat. Methods.7:248–249.
Arpaia E., Shahar M., Dadi H., Cohen A., and Roifman C.M.1994. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell. 76:947–958.
Atkinson A.R. and Roifman C.M.2007. Low serum immunoglobulin G2 levels in infancy can be transient. Pediatrics. 120:e543–e547.
Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., and Abecasis G.R.2015. A global reference for human genetic variation. Nature. 526:68–74.
Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., and Shendure J.2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet.12:745–755.
Blake J.A., Dolan M., Drabkin H., Hill D.P., Li N., Sitnikov D., Bridges S., Burgess S., Buza T., Mccarthy F., Peddinti D., Pillai L., Carbon S., Dietze H., Ireland A., Lewis S.E., Mungall C.J., Gaudet P., Chrisholm R.L., Fey P., Kibbe W.A., Basu S., Siegele D.A., Mcintosh B.K., Renfro D.P., Zweifel A.E., Hu J.C., Brown N.H., Tweedie S., Alam-Faruque Y., Apweiler R., Auchinchloss A., Axelsen K., Bely B., Blatter M., Bonilla C., Bouguerleret L., Boutet E., Breuza L., Bridge A., Chan W.M., Chavali G., Coudert E., Dimmer E., Estreicher A., Famiglietti L., Feuermann M., Gos A., Gruaz-Gumowski N., Hieta R., Hinz C., Hulo C., Huntley R., James J., Jungo F., Keller G., Laiho K., Legge D., Lemercier P., Lieberherr D., Magrane M., Martin M.J., Masson P., Mutowo-Muellenet P., O’Donovan C., Pedruzzi I., Pichler K., Poggioli D., Porras Millan P., Poux S., Rivoire C., Roechert B., Sawford T., Schneider M., Stutz A., Sundaram S., Tognolli M., Xenarios I., Foulgar R., Lomax J., Roncaglia P., Khodiyar V.K., Lovering R.C., Talmud P.J., Chibucos M., Giglio M.G., Chang H., Hunter S., Mcanulla C., Mitchell A., Sangrador A., Stephan R., Harris M.A., Oliver S.G., Rutherford K., Wood V., Bahler J., Lock A., Kersey P.J., McDowall D.M., Staines D.M., Dwinell M., Shimoyama M., Laulederkind S., Hayman T., Wang S.-J., Petri V., Lowry T., D’Eustachio P., Matthews L., Balakrishnan R., Binkley G., Cherry J.M., Costanzo M.C., Dwight S.S., Engel S.R., Fisk D.G., Hitz B.C., Hong E.L., Karra K., Miyasato S.R., Nash R.S., Park J., Skrzypek M.S., Weng S., Wong E.D., Berardini T.Z., Huala E., Mi H., Thomas P.D., Chan J., Kishore R., Sternberg P., Van Auken K., Howe D., and Westerfield M.2013. Gene Ontology annotations and resources. Nucleic Acids Res.41:D530–D535.
Buck D., Moshous D., de Chasseval R., Ma Y., le Deist F., Cavazzana-Calvo M., Fischer A., Casanova J.L., Lieber M.R., and de Villartay J.P.2006. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur. J. Immunol.36:224–235.
Dadi H.K., Simon A.J., and Roifman C.M.2003. Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. N. Engl. J. Med.349:1821–1828.
de Miranda N.F., Bjorkman A., and Pan-Hammarstrom Q.2011. DNA repair: The link between primary immunodeficiency and cancer. Ann. N.Y. Acad. Sci.1246:50–63.
Enders A., Fisch P., Schwarz K., Duffner U., Pannicke U., Nikolopoulos E., Peters A., Orlowska-Volk M., Schindler D., Friedrich W., Selle B., Niemeyer C., and Ehl S.2006. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J. Immunol.176:5060–5068.
Eppig J.T., Blake J.A., Bult C.J., Kadin J.A., and Richardson J.E.2015. The Mouse Genome Database (MGD): Facilitating mouse as a model for human biology and disease. Nucleic Acids Res.43:D726–D736.
Erikson G.A., Bodian D.L., Rueda M., Molparia B., Scott E.R., Scott-Van Zeeland A.A., Topol S.E., Wineinger N.E., Niederhuber J.E., Topol E.J., and Torkamani A.2016. Whole-genome sequencing of a healthy aging cohort. Cell. 165:1002–1011.
Fabregat A., Sidiropoulos K., Garapati P., Gillespie M., Hausmann K., Haw R., Jassal B., Jupe S., Korninger F., Mckay S., Matthews L., May B., Milacic M., Rothfels K., Shamovsky V., Webber M., Weiser J., Williams M., Wu G., Stein L., Hermjakob H., and D’Eustachio P.2016. The reactome pathway knowledgebase. Nucleic Acids Res.44:D481–D487.
Gruhn B., Seidel J., Zintl F., Varon R., Tonnies H., Neitzel H., Bechtold A., Hoehn H., and Schindler D.2007. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet. J. Rare Dis.2:5.
Grunebaum E., Bates A., and Roifman C.M.2008. Omenn syndrome is associated with mutations in DNA ligase IV. J. Allergy Clin. Immunol.122:1219–1220.
Hayani A., Suarez C.R., Molnar Z., LeBeau M., and Godwin J.1994. Acute myeloid leukaemia in a patient with Seckel syndrome. J. Med. Genet.31:148–149.
IJspeert H., Warris A., van der Flier M., Reisli I., Keles S., Chishimba S., van Dongen J.J., van Gent D.C., and van der Burg M.2013. Clinical spectrum of LIG4 deficiency is broadened with severe dysmaturity, primordial dwarfism, and neurological abnormalities. Hum. Mutat.34:1611–1614.
Kanehisa M., Sato Y., Kawashima M., Furumichi M., and Tanabe M.2016. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res.44:D457–D462.
Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., and Shendure J.2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet.46:310–315.
Kohler S., Doelken S.C., Mungall C.J., Bauer S., Firth H.V., Bailleul-Forestier I., Black G.C., Brown D.L., Brudno M., Campbell J., Fitzpatrick D.R., Eppig J.T., Jackson A.P., Freson K., Girdea M., Helbig I., Hurst J.A., Jahn J., Jackson L.G., Kelly A.M., Ledbetter D.H., Mansour S., Martin C.L., Moss C., Mumford A., Ouwehand W.H., Park S.M., Riggs E.R., Scott R.H., Sisodiya S., Van Vooren S., Wapner R.J., Wilkie A.O., Wright C.F., Vulto-van Silfhout A.T., de Leeuw N., de Vries B.B., Washingthon N.L., Smith C.L., Westerfield M., Schofield P., Ruef B.J., Gkoutos G.V., Haendel M., Smedley D., Lewis S.E., and Robinson P.N.2014. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res.42:D966–D974.
Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J., Jang W., Katz K., Ovetsky M., Riley G., Sethi A., Tully R., Villamarin-Salomon R., Rubinstein W., and Maglott D.R.2016. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res.44:D862–D868.
Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., Tukiainen T., Birnbaum D.P., Kosmicki J.A., Duncan L.E., Estrada K., Zhao F., Zou J., Pierce-Hoffman E., Berghout J., Cooper D.N., Deflaux N., DePristo M., Do R., Flannick J., Fromer M., Gauthier L., Goldstein J., Gupta N., Howrigan D., Kiezun A., Kurki M.I., Moonshine A.L., Natarajan P., Orozco L., Peloso G.M., Poplin R., Rivas M.A., Ruano-Rubio V., Rose S.A., Ruderfer D.M., Shakir K., Stenson P.D., Stevens C., Thomas B.P., Tiao G., Tusie-Luna M.T., Weisburd B., Won H.H., Yu D., Altshuler D.M., Ardissino D., Boehnke M., Danesh J., Donnelly S., Elosua R., Florez J.C., Gabriel S.B., Getz G., Glatt S.J., Hultman C.M., Kathiresan S., Laakso M., McCarroll S., McCarthy M.I., McGovern D., McPherson R., Neale B.M., Palotie A., Purcell S.M., Saleheen D., Scharf J.M., Sklar P., Sullivan P.F., Tuomilehto J., Tsuang M.T., Watkins H.C., Wilson J.G., Daly M.J., and MacArthur D.G.2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536:285–291.
Marrella V., Poliani P.L., Notarangelo L.D., Grassi F., and Villa A.2014. Rag defects and thymic stroma: Lessons from animal models. Front. Immunol.5:259.
Merico D.2016. Whole exome and genome sequencing for Mendelian immune disorders: From molecular diagnostics to new disease variant and gene discovery. LymphoSign Journal. 3(4): 135–158.
Murray J.E., Bicknell L.S., Yigit G., Duker A.L., van Kogelenberg M., Haghayegh S., Wieczorek D., Kayserili H., Albert M.H., Wise C.A., Brandon J., Kleefstra T., Warris A., van der Flier M., Bamforth J.S., Doonanco K., Ades L., Ma A., Field M., Johnson D., Shackley F., Firth H., Woods C.G., Nurnberg P., Gatti R.A., Hurles M., Bober M.B., Wollnik B., and Jackson A.P.2014. Extreme growth failure is a common presentation of ligase IV deficiency. Hum. Mutat.35:76–85.
Ng P.C. and Henikoff S.2001. Predicting deleterious amino acid substitutions. Genome Res.11:863–874.
O’Driscoll M., Cerosaletti K.M., Girard P.M., Dai Y., Stumm M., Kysela B., Hirsch B., Gennery A., Palmer S.E., Seidel J., Gatti R.A., Varon R., Oettinger M.A., Neitzel H., Jeggo P.A., and Concannon P.2001. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell.8:1175–1185.
Plowman P.N., Bridges B.A., Arlett C.F., Hinney A., and Kingston J.E.1990. An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. Br. J. Radiol.63:624–628.
Poliani P.L., Facchetti F., Ravanini M., Gennery A.R., Villa A., Roifman C.M., and Notarangelo L.D.2009. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: Possible implications for the pathophysiology of Omenn syndrome. Blood. 114:105–108.
Pollard K.S., Hubisz M.J., Rosenbloom K.R., and Siepel A.2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res.20:110–121.
Ratech H., Greco M.A., Gallo G., Rimoin D.L., Kamino H., and Hirschhorn R.1985. Pathologic findings in adenosine deaminase-deficient severe combined immunodeficiency. I. Kidney, adrenal, and chondro-osseous tissue alterations. Am. J. Pathol.120:157–169.
Reva B., Antipin Y., and Sander C.2011. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res.39:e118.
Riballo E., Critchlow S.E., Teo S.H., Doherty A.J., Priestley A., Broughton B., Kysela B., Beamish H., Plowman N., Arlett C.F., Lehmann A.R., Jackson S.P., and Jeggo P.A.1999. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol.9:699–702.
Roifman C.M.2005. Studies of patients’ thymi aid in the discovery and characterization of immunodeficiency in humans. Immunol. Rev.203:143–155.
Roifman C.M., Gu Y., and Cohen A.2006. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome. J. Allergy Clin. Immunol.117:897–903.
Roifman C.M., Hummel D., Martinez-Valdez H., Thorner P., Doherty P.J., Pan S., Cohen F., and Cohen A.1989. Depletion of CD8+ cells in human thymic medulla results in selective immune deficiency. J. Exp. Med.170:2177–2182.
Rucci F., Poliani P.L., Caraffi S., Paganini T., Fontana E., Giliani S., Alt F.W., and Notarangelo L.D.2011. Abnormalities of thymic stroma may contribute to immune dysregulation in murine models of leaky severe combined immunodeficiency. Front. Immunol.2:pii: 00015.
Sharfe N., Dadi H.K., Shahar M., and Roifman C.M.1997. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA.94:3168–3171.
Sharfe N., Merico D., Karanxha A., Macdonald C., Dadi H., Ngan B., Herbrick J.A., and Roifman C.M.2015. The effects of RelB deficiency on lymphocyte development and function. J. Autoimmun.65:90–100.
Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., Weinstock G.M., Wilson R.K., Gibbs R.A., Kent W.J., Miller W., and Haussler D.2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res.15:1034–1050.
Slatter M.A. and Gennery A.R.2010. Primary immunodeficiencies associated with DNA-repair disorders. Expert Rev. Mol. Med.12:e9.
Smith C.L. and Eppig J.T.2012. The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome.23:653–668.
Solomon B.D., Nguyen A.D., Bear K.A., and Wolfsberg T.G.2013. Clinical genomic database. Proc. Natl. Acad. Sci. USA.110:9851–9855.
Stavropoulos D.J., Merico D., Jobling R., Bowdin S., Monfared N., Thiruvahindrapuram B., Nalpathamkalam T., Pellecchia G., Yuen R.K.C., Szego M.J., Hayeems R.Z., Shaul R.Z., Brudno M., Girdea M., Frey B., Alipanahi B., Ahmed S., Babul-Hirji R., Porras R.B., Carter M.T., Chad L., Chaudhry A., Chitayat D., Doust S.J., Cytrynbaum C., Dupuis L., Ejaz R., Fishman L., Guerin A., Hashemi B., Helal M., Hewson S., Inbar-Feigenberg M., Kannu P., Karp N., Kim R.H., Kronick J., Liston E., MacDonald H., Mercimek-Mahmutoglu S., Mendoza-Londono R., Nasr E., Nimmo G., Parkinson N., Quercia N., Raiman J., Roifman M., Schulze A., Shugar A., Shuman C., Sinajon P., Siriwardena K., Weksberg R., Yoon G., Carew C., Erickson R., Leach R.A., Klein R., Ray P.N., Meyn M.S., Scherer S.W., Cohn R.D., and Marshall C.R.2016. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom. Med.1:15012.
Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A., and Cooper D.N.2014. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet.133:1–9.
Tammimies K., Marshall C.R., Walker S., Kaur G., Thiruvahindrapuram B., Lionel A.C., Yuen R.K., Uddin M., Roberts W., Weksberg R., Woodbury-Smith M., Zwaigenbaum L., Anagnostou E., Wang Z., Wei J., Howe J.L., Gazzellone M.J., Lau L., Sung W.W., Whitten K., Vardy C., Crosbie V., Tsang B., D’Abate L., Tong W.W., Luscombe S., Doyle T., Carter M.T., Szatmari P., Stuckless S., Merico D., Stavropoulos D.J., Scherer S.W., and Fernandez B.A.2015. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 314:895–903.
Toita N., Hatano N., Ono S., Yamada M., Kobayashi R., Kobayashi I., Kawamura N., Okano M., Satoh A., Nakagawa A., Ohshima K., Shindoh M., Takami T., Kobayashi K., and Ariga T.2007. Epstein-Barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. Am. J. Med. Genet. A.143A:742–745.
Unal S., Cerosaletti K., Uckan-Cetinkaya D., Cetin M., and Gumruk F.2009. A novel mutation in a family with DNA ligase IV deficiency syndrome. Pediatr. Blood Cancer. 53:482–484.
van der Burg M., van Veelen L.R., Verkaik N.S., Wiegant W.W., Hartwig N.G., Barendregt B.H., Brugmans L., Raams A., Jaspers N.G., Zdzienicka M.Z., van Dongen J.J., and van Gent D.C.2006. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J. Clin. Invest.116:137–145.
Wang K., Li M., and Hakonarson H.2010. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res.38:e164.
Xiong H.Y., Alipanahi B., Lee L.J., Bretschneider H., Merico D., Yuen R.K., Hua Y., Gueroussov S., Najafabadi H.S., Hughes T.R., Morris Q., Barash Y., Krainer A.R., Jojic N., Scherer S.W., Blencowe B.J., and Frey B.J.2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 347:1254806.
Yang K., Guo R., and Xu D.2016. Non-homologous end joining: Advances and frontiers. Acta Biochim. Biophys. Sin. (Shanghai).48:632–640.
Yano M., Kuroda N., Han H., Meguro-Horike M., Nishikawa Y., Kiyonari H., Maemura K., Yanagawa Y., Obata K., Takahashi S., Ikawa T., Satoh R., Kawamoto H., Mouri Y., and Matsumoto M.2008. Aire controls the differentiation program of thymic epithelial cells in the medulla for the establishment of self-tolerance. J. Exp. Med.205:2827–2838.
Yue J., Lu H., Lan S., Liu J., Stein M.N., Haffty B.G., and Shen Z.2013. Identification of the DNA repair defects in a case of Dubowitz syndrome. PLoS ONE. 8:e54389.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 1 • March 2017
Pages: 31 - 41
History
Received: 26 January 2017
Accepted: 2 March 2017
Accepted manuscript online: 2 March 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
WillaLiao, BoNgan, DanieleMerico, HarjitDadi, and Chaim M.Roifman. 2017. A novel mutation in LIG4 in an infant presenting with severe combined immunodeficiency with thymic medullary dysplasia. LymphoSign Journal.
4(1): 31-41. https://doi.org/10.14785/lymphosign-2017-0001
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
