Immunodeficiency and severe gastrointestinal manifestations in a patient with novel DKC1 mutations causing Hoyeraal–Hreidarsson syndrome
Abstract
Background: Hoyeraal–Hreidarsson syndrome (HHS) is considered a clinically severe variant of dyskeratosis congenita (DKC) and represents the extreme phenotype caused by aberrant telomere biology. Unlike patients with DKC who present later in life, most cases of HHS present in the first years of life. Clinical features include intrauterine growth restriction and microcephaly, which are universal but not pathognomonic, as well as gastrointestinal, immunological and neurological manifestations. The immunological profile is varied as a result of cellular immunodeficiency, humoral defects, or both, and may be the presenting symptom of these patients. Moreover, the immunological phenotype can change over time, making HHS a diagnostic challenge.
Methods: This case report highlights the clinical presentation and immune investigations of a male patient with a novel mutation in DKC1, causing HHS.
Results: Here, we describe a patient with HHS who presented with Pneumocystis jiroveci pneumonia and low T cells, which is typical of severe combined immunodeficiency. Over time, he developed agammaglobulinemia whereas T-cell function improved. He also presented with extremely severe gastrointestinal manifestations, and died at 3 years of age.
Conclusion: This case report highlights a novel compound heterozygous mutation in DKC1, and the need to consider HHS as the differential diagnosis of patients with combined immunodeficiency.
Statement of novelty: The case reports on a novel mutation in DKC1.
Introduction
Hoyeraal–Hreidarsson syndrome (HHS) is considered a clinically severe variant of dyskeratosis congenita (DKC) and represents the extreme phenotype caused by aberrant telomere biology (Knight et al. 1999; Kirwan and Dokal 2008; Dokal 2011; Touzot et al. 2012; Glousker et al. 2015). It is a multisystem genetic disorder typically caused by germline mutations in telomere biology genes (Glousker et al. 2015). Very short (<first percentile for age) leukocyte telomere lengths are diagnostic of DKC and HHS (Marrone et al. 2007; Glousker et al. 2015). Patients present early in childhood with the constellation of cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure (BMF), and intrauterine growth retardation (IUGR), which comprise the majority of the clinical complications. The DKC-associated mucocutaneous triad of nail dysplasia, lacy skin pigmentation, and oral leukoplakia may be present at diagnosis or develop over time in patients with HHS. The eponym HHS was first proposed by a 1995 case report describing a child presenting with progressive pancytopenia, cerebellar hypoplasia, prenatal growth retardation, microcephaly, and developmental delay (Knight et al. 1999). These clinical features were noted as strikingly similar to the clinical description of the patients reported by Hoyeraal et al. (1970) and Hreidarsson et al. (1988). About 50 cases of HHS have been reported (Berthet et al. 1995; Knight et al. 1999; Revy et al. 2000; Cossu et al. 2002; Lamm et al. 2009; Touzot et al. 2012; Ballew et al. 2013; Deng et al. 2013; Le Guen et al. 2013; Kocak et al. 2014; Malbora et al. 2014). To date, X-linked recessive mutations in DKC1 (encoding dyskerin), autosomal dominant mutations in TINF2 (encoding TIN2, also termed TINF2), and autosomal recessive mutations in TERT, ACD (encoding TPP1, also termed ACD), and RTEL1, have been associated with the syndrome (Kirwan and Dokal 2008; Le Guen et al. 2013; Glousker et al. 2015). All HHS-associated genes encode proteins with specialized telomeric functions: TERT and dyskerin are components of the telomerase ribonucleoprotein complex (Kirwan and Dokal 2008), TIN2 and TPP1 are components of the telomeric shelterin complex (Kirwan and Dokal 2008), and RTEL1 is a helicase important in telomere biology (Le Guen et al. 2013). However, dyskerin, RTEL1, and TERT have also been reported to have nontelomeric functions syndrome (Kirwan and Dokal 2008; Le Guen et al. 2013; Glousker et al. 2015). Therefore, the question remains whether nontelomeric defects contribute to the pathology of HHS, perhaps distinguishing it from DKC.
Gastrointestinal involvement has been documented in most reported cases and poses a significant challenge in the supportive treatment of HHS (Sznajer et al. 2003; Borggraefe et al. 2009; Touzot et al. 2012). Initially, infants with HHS and IUGR may present with feeding difficulties due to delayed development. Oesophageal strictures, sometimes requiring repetitive endoscopic dilations, are present in about 25% of reported cases (Borggraefe et al. 2009). Severe enteropathy, manifesting as protracting noninfectious diarrhea, is also reported in another quarter of cases (Borggraefe et al. 2009). Intestinal biopsies may show a change in the proliferative compartment of the mucosa, leading to glandular atrophy (Sznajer et al. 2003). Colitis has also been described with non-specific lymphoplasmocytic infiltrate and chronic edema, as well as granulomatous lesions associated with mononuclear and eosinophilic infiltrate (Borggraefe et al. 2009; Touzot et al. 2012).
Immunodeficiency is an under-recognized feature of HHS. Patients are reported to have a progressive immune deficiency, manifesting as an increased susceptibility to life-threatening infections. Lymphopenia is the most common immunological abnormality observed in this syndrome (approximately 56% of patients). A decreased count of B cells and NK cells is often the most remarkable feature. In particular, a virtual absence of B lymphocytes from birth has been observed (Hoyeraal et al. 1970; Hreidarsson et al. 1988; Berthet et al. 1995; Jullien et al. 2016; Cossu et al. 2002; Touzot et al. 2010; Jyonouchi et al. 2011; Touzot et al. 2012; Le Guen et al. 2013). This results in hypogammaglobulinemia and impaired antibody production in response to vaccination. The marked involvement of the B-cell compartment is likely due to the additional cell proliferation that B lymphocytes undergo during their development (thus resulting in accelerated telomere shortening, see below), coupled with a shorter life span as compared with T lymphocytes (Hodes and Hathcock 2002). The T-cell compartment appears to be less frequently affected, with 16% of reported patients exhibiting a decrease in T cell counts (CD4 and (or) CD8 counts). Abnormalities of T-cell proliferation in response to specific antigens (candida and tetanus) and less frequently to mitogens have also been observed, indicating reduced T-cell function (Sznajer et al. 2003; Ballew et al. 2013). Some cases presenting as severe combined immunodeficiency (SCID) have also been reported (Cossu et al. 2002). Thus, the diagnosis of HHS should be considered in any child presenting with agammaglobulinemia or combined immunodeficiency associated with neurological features (such as microcephaly and (or) cerebellar hypoplasia).
We present a patient with a lethal novel mutation in the DKC1 gene who presented with Pneumocystis jiroveci pneumonia (PJP), SCID-like phenotype, and severe gastrointestinal manifestations.
Case presentation
The patient was born to healthy parents of East Indian descent with no history of consanguinity. He was conceived in India in a fifth attempt of artificial insemination of which the father was the donor. Mother was a 33-year-old G1, P0. Family history was unremarkable. The pregnancy course was complicated by severe hyperemesis gravidarum treated with Diclectin and metoclopramide. HIV serology was negative. An ultrasound done at the 34th week of gestation showed symmetric IUGR. The patient was born at weighing 1200 g at 34 weeks of gestation via cesarean section due to fetal bradycardia, which was attributed to uteroplacental insufficiency. He was short but his parents were also at the third percentile for height. He remained hospitalized for 6 weeks for feeding and difficulties. He developed Candida diaper rash in the NICU and was successfully treated with antifungal cream. He was subsequently discharged home with a diagnosis of failure-to-thrive (FTT), to be followed by his family physician, and anemia, which resolved at 4 months.
At 5.5 months of age he was hospitalized due to respiratory distress. Upon his admission his chest x-ray showed patchy infiltrates bilaterally. P. jiroveci was isolated from bronchoalveolar lavage, and Septra in combination with steroid treatment was initiated. He deteriorated and needed assisted ventilation for 15 days. His initial blood count revealed normal leukocyte counts 11.8 (6.0–17.5 109/L), hemoglobin 126 (95–135 g/L), platelets 437 (150–400 × 109/L), neutrophils 9.40 (1.00–8.50 × 109/L), but low lymphocytes 1.30 (4.00–13.50 × 109/L). Serum albumin was low at 25 g/L (N 35–50), but electrolytes, liver function, renal function, and coagulation tests were all normal. In light of his hypogammaglobulinemia, IVIG treatment was initiated. Immunological assessments performed at 6 and 8 months of age are shown in Table 1. Whereas mitogen proliferation studies were normal, TREC levels were low. Colitis developed at 6–12 months of age with mucoid stools and specks of blood. A colon biopsy was performed showing marked disruption in normal crypt architecture with areas of crypt dropout. There was mild to moderate inflammatory infiltrate in the lamina propria, consisting mostly of neutrophils and occasional eosinophils, a paucity of lymphocytes and an absence of plasma cells. A rectal biopsy showed inflammatory exudate and focal crypt abscesses and erosions. Viral staining, cultures, electron microscopy, and C. difficile were negative. No response to broad spectrum antibiotic coverage was noted. Due to neutropenia, a bone marrow biopsy revealed an arrest of megakaryocyte maturation. Other lineages showed all stages of normal maturation.
Table 1:
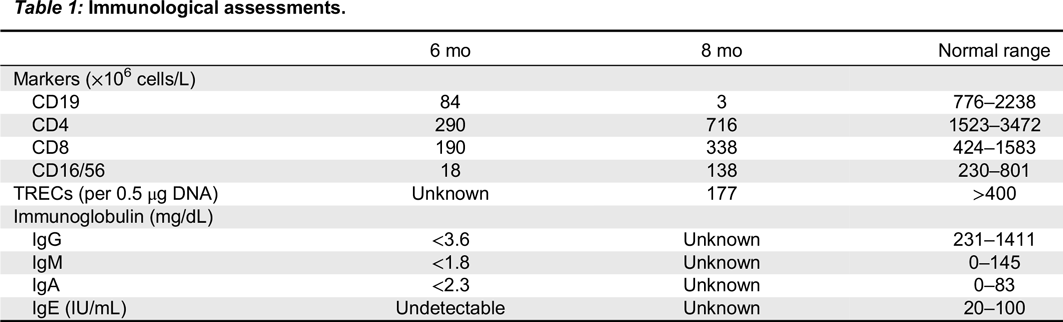
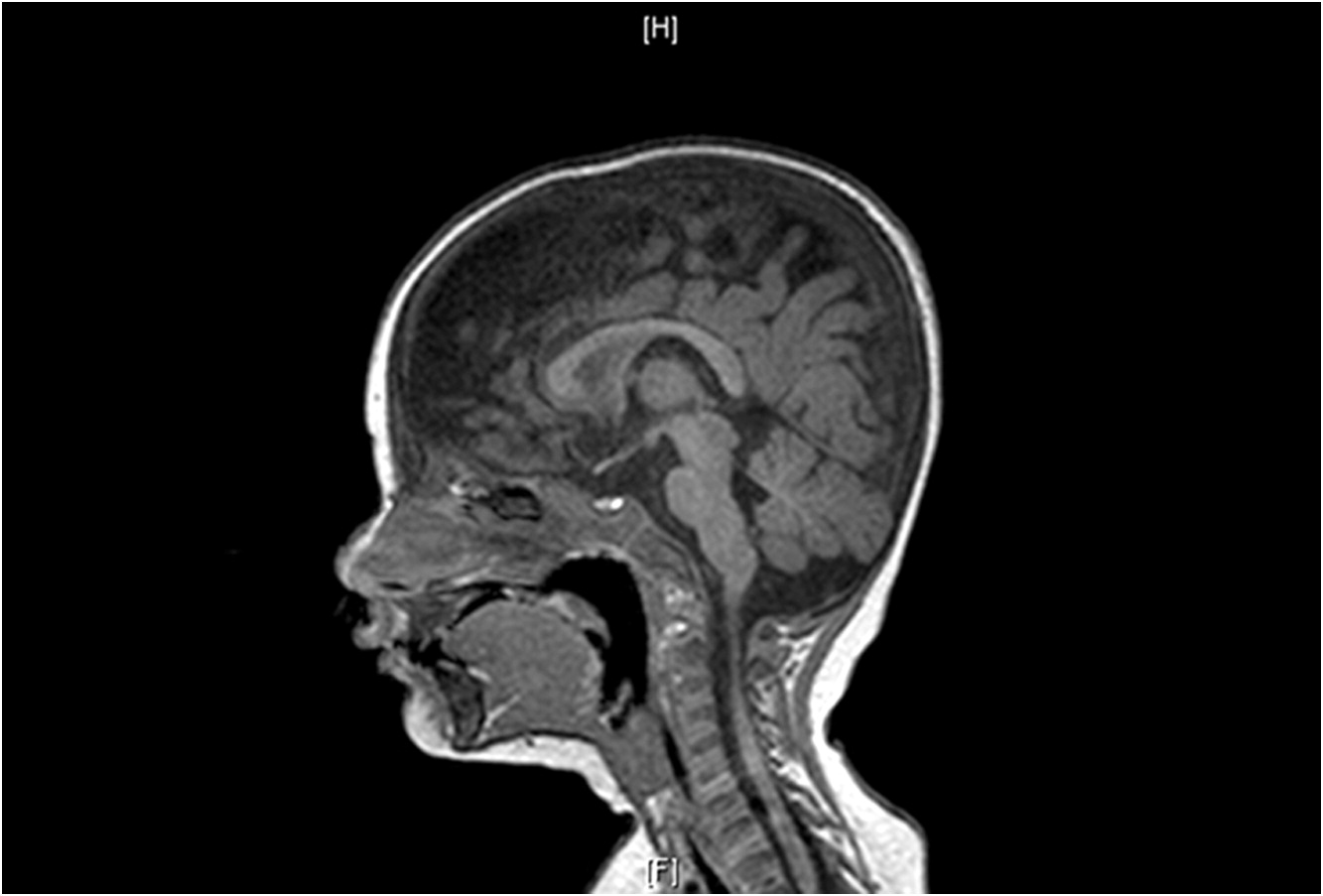
At 1 year of age when charting the patient’s growth parameters, it became apparent that his head circumference was far below the 3rd centile, and disproportionally small when compared with other growth parameters (Figures 1 and 2). This prompted an MRI of the brain which showed prominent extra-cerebral CSF spaces and mild diffuse cerebellar volume loss (Figure 3). Hyperpigmented lesions also appeared.
Figure 1:
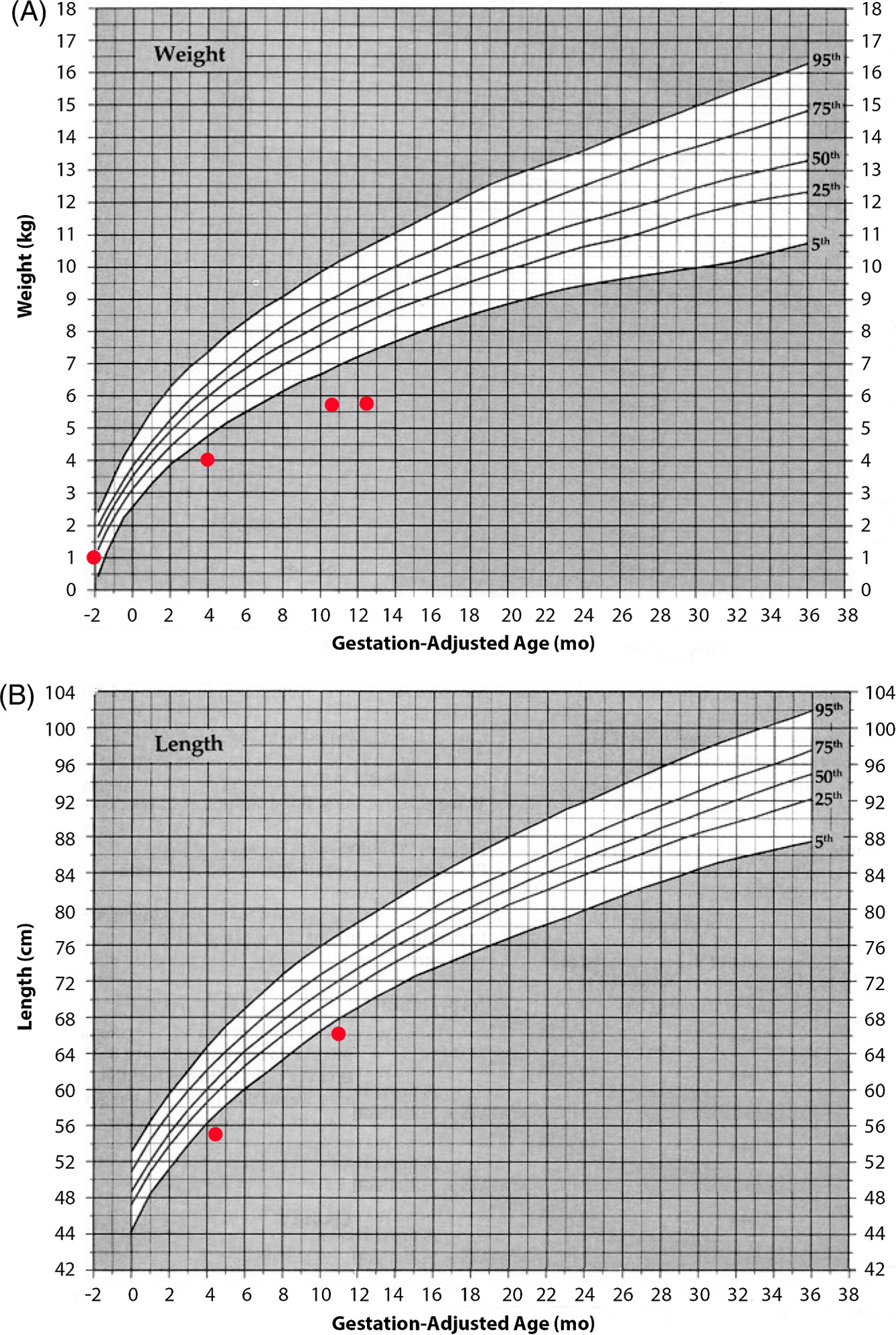
Figure 2:
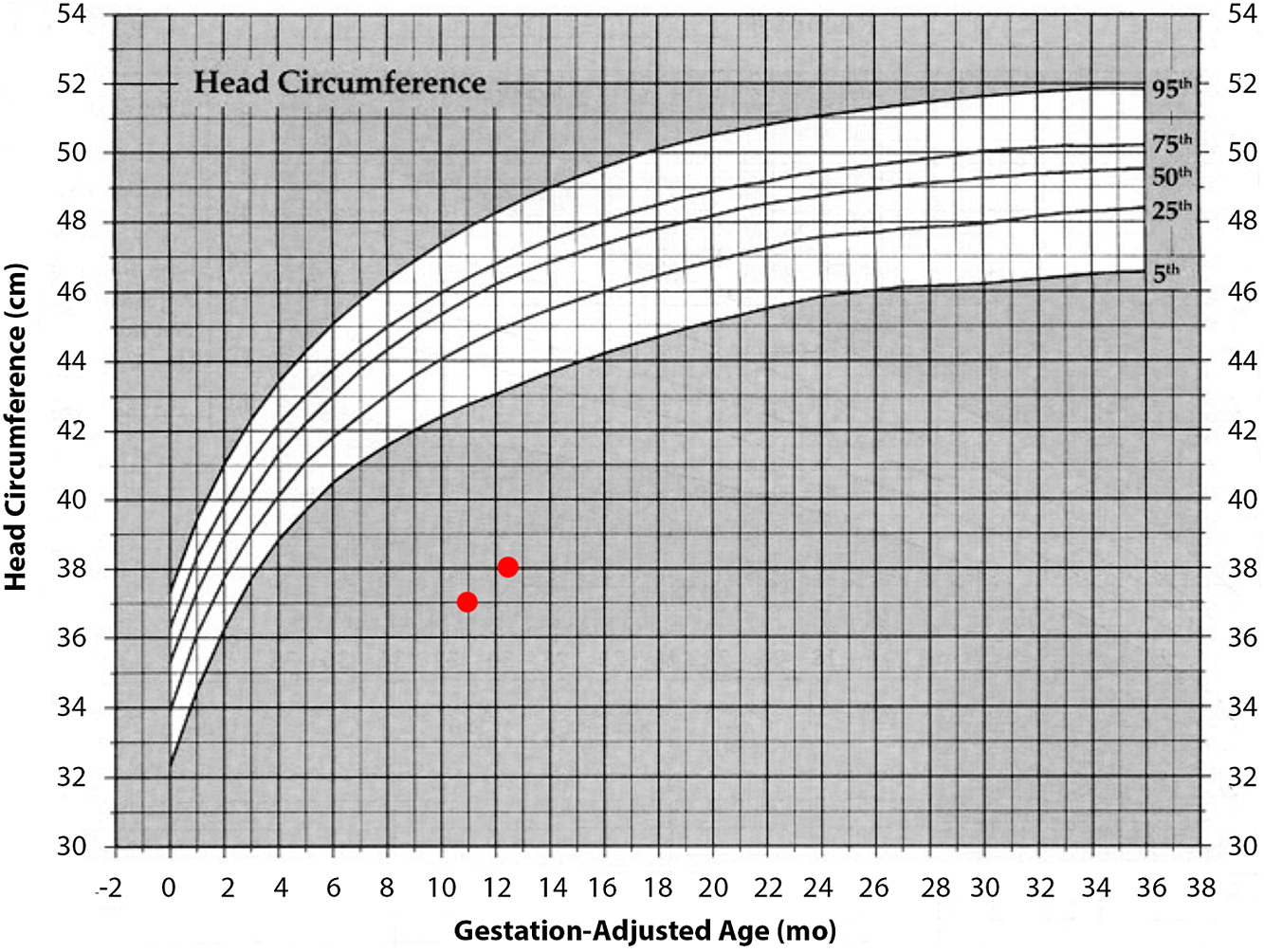
Figure 3:
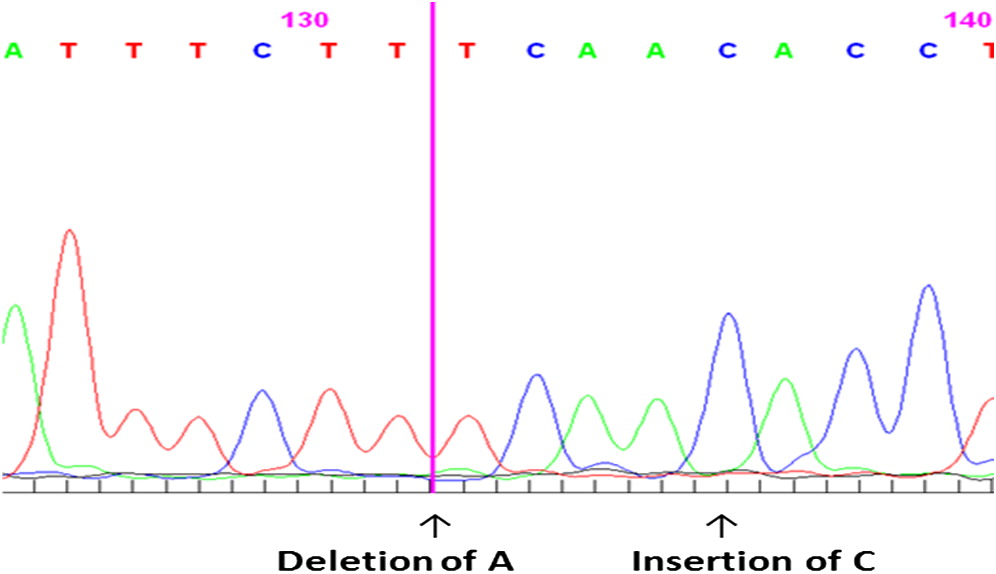
Although SCID panel gene sequencing did not detect any abnormalities, DKC1 gene sequencing revealed a novel mutation, thus the diagnosis of HHS was established. Genetic workup by Sanger sequencing revealed compound heterozygosity for a DKC1 gene mutation. We identified a novel mutation g.2601-2602 insertion C, resulting in an amino acid change K39T, and mutation g.2597 deletion A, resulting in an amino acid change I38S (Figure 4).
Figure 4:
The patient became TPN dependent because of severe diarrhea and protein losing enteropathy. He was not growing well and suffered from electrolyte imbalance. Neurologic manifestations included head circumference arrest and significant developmental delay. Ataxia also manifested as a broad-based gait. His clinical course was complicated by severe infections and ICU admissions. Thrombocytopenia and anemia developed, and at 3 years of age, he died of liver, renal, and respiratory failure with severe hyperammonemia. The family denied an autopsy.
Discussion
HHS is a rare form of DKC characterized by clinical features that include IUGR and microcephaly, which are universal. Other features include ataxia, and skin and mucus membrane changes. All patients eventually develop BMF. The age of onset varies between 1–77 months of age, with most cases presenting at 1–2 years. The patient presented with IUGR and microcephaly, but because of his prematurity and FTT, the extent and severity of his microcephaly was only noticed when he developed complete arrest of his head circumference. The involvement of the immune system has been reported to be variable in patients with HHS, and decreased numbers of T, B, or NK cells have been described with an inconsistent pattern changing over time. B cells are more commonly affected but patients can present with a SCID-like phenotype.
These findings can precede signs of BMF, thus highlighting the need to consider HHS as the deferential diagnosis of patients with combined immunodeficiency. In particular, the patient presented with a SCID-like presentation with PJP and low T-cell numbers. Over time, the T-cell numbers and function recovered. His B-cell numbers, however, remained extremely low and he was dependent on IVIG treatment due to agammaglobulinemia. Because of his severe gastrointestinal manifestations causing protein losing enteropathy, he eventually needed IVIG replacement therapy every 2 weeks.
Gastrointestinal involvement in patients with HHS is common and can include oral ulcerations, esophageal dysmotility with stenosis, and severe diarrhea. Diarrhea was reported in 52% (13 of 25) of all described patients in the literature (Borggraefe et al. 2009). The mean age of onset of diarrhea was 12 months. In 7 patients (28%) diarrhea was the initial complaint of the disease (Borggraefe et al. 2009), suggesting that inflammation of the gut is a consistent and early finding in HHS. In this patient, gastrointestinal involvement was noted from 5 months of age and was his major cause of morbidity, with sloughing of the GI tract. A thorough infectious work-up was undertaken but all cultures were negative. A rectal biopsy showed inflammatory exudate and focal crypt abscesses and erosions. The extremely severe gastrointestinal presentation was attributed to the short life span of the enterocytes, due to telomere abnormalities. He eventually did not tolerate oral feeding and was TPN dependent. Based on one report of improvement after steroid treatment, he was given a trial of methylprednisolone treatment with a low dose of 0.8 mg/kg/day for 2 weeks. He did not show any improvement and the treatment was therefore discontinued.
No skin mucus membranes or nail anomalies were initially seen in the patient until he was 1 year of age. He developed mild hyperpigmented lesions and at 2.5 years developed leukoplakia. He did not develop ataxia until 2.5 years of age. Almost all HHS patients die before 4 years of age, although those with a milder form have been reported to survive longer (Ozdemir et al. 2004). In this case, while the patient had an extremely severe and early onset, which needed intensive supportive treatment, he survived to 3 years of age but died of multiple organ failure. In other patients the reported main cause of death is infection.
Unlike other SCIDs for which early bone marrow transplantation is beneficial, curative treatment for HHS remains elusive.
Conclusion
We present here a patient with HHS with a novel genetic mutation. His clinical presentation was due to immunodeficiency with a SCID-like phenotype, with low CD4 counts and PJP. This progressed to a complete lack of B cells and agammaglobulinemia; however, T-cell numbers and function improved. His other main clinical presentation was an extremely severe diarrhea that presented at a very early age.
This case should alert clinicians to the possibility that HHS can present with a SCID phenotype and severe gastrointestinal disease as the symptoms of this syndrome. These occur before typical signs of BMF, ataxia, or skin and mucus membranes appear. Consequently, molecular diagnosis should be pursued before a decision to proceed to bone marrow transplantation is made.
REFERENCES
Aalfs C.M., van den Berg H., Barth P.G., and Hennekam R.C.1995. The Hoyeraal-Hreidarsson syndrome: the fourth case of a separate entity with prenatal growth retardation, progressive pancytopenia and cerebellar hypoplasia. Eur J Pediatr.154(4):304–308.
Ballew B.J., Joseph V., De S., Sarek G., Vannier J.B., Stracker T., Schrader K.A., Small T.N., O’Reilly R., Manschreck C., Harlan Fleischut M.M., Zhang L., Sullivan J., Stratton K., Yeager M., Jacobs K., Giri N., Alter B.P., Boland J., Burdett L., Offit K., Boulton S.J., Savage S.A., and Petrini J.H.2013. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet.9(8):e1003695.
Berthet F., Tuchschmid P., Boltshauser E., and Seger R.A.1995. The Hoyeraal-Hreidarsson syndrome: don’t forget the associated immunodeficiency. Eur J Pediatr.154(12):998.
Borggraefe I., Koletzko S., Arenz T., Fuehrer M., Hoffmann F., Dokal I., Vulliamy T., Weiler V., Griese M., Belohradsky B.H., and Lang T.2009. Severe variant of x-linked dyskeratosis congenita (Hoyeraal-Hreidarsson Syndrome) causes significant enterocolitis in early infancy. Pediatr Gastroenterol Nutr.49(3):359–363.
Cossu F., Vulliamy T.J., Marrone A., Badiali M., Cao A., and Dokal I.2002. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br J Haematol.119(3):765–768.
Deng Z., Glousker G., Molczan A., Fox A.J., Lamm N., Dheekollu J., Weizman O.E., Schertzer M., Wang Z., Vladimirova O., Schug J., Aker M., Londoño-Vallejo A., Kaestner K.H., Lieberman P.M., and Tzfati Y.2013. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA.110(36):E3408–E3416.
Dokal I.2011. Dyskeratosis congenital. Hematology Am Soc Hematol Educ Program.2011:480–486.
Glousker G., Touzot F., Revy P., Tzfati Y., and Savage S.A.2015. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol.170(4):457–471.
Hodes R.J., Hathcock K.S., and Weng N.P. 2002. Telomeres in T and B cells. Nat Rev Immunol.2(9):699–706.
Hoyeraal H.M., Lamvik J., and Moe P.J.1970. Congenital hypoplastic thromboycytopenia and cerebral malformations in two brothers. Acta Paediatr Scand.59(2): 185–191.
Hreidarsson S., Kristjansson K., Johannesson G., and Johannsson J.H.1988. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr Scand.77(5): 773–775.
Jullien L., Kannengiesser C., Kermasson L., Cormier-Daire V., Leblanc T., Soulier J., Londono-Vallejo A., de Villartay J.P., Callebaut I., and Revy P.2016. Mutations of the RTEL1 helicase in a hoyeraal-hreidarsson syndrome patient highlight the importance of the ARCH domain. Hum Mutat.37(5):469–472.
Jyonouchi S., Forbes L., Ruchelli E., and Sullivan K.E.2011. Dyskeratosis congenita: a combined immunodeficiency with broad clinical spectrum — a single-center pediatric experience. Pediatr Allergy Immu22:313–319.
Kirwan M. and Dokal I.2008. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet.73(2):103–112.
Knight S.W., Heiss N.S., Vulliamy T.J., Aalfs C.M., McMahon C., Richmond P., Jones A., Hennekam R.C., Poustka A., Mason P.J., and Dokal I.1999. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1S. Br J Haematol.107(2):335–339.
Kocak H., Ballew B.J., Bisht K., Eggebeen R., Hicks B.D., Suman S., O’Neil A., and Giri N., NCI DCEG Cancer Genomics Research Laboratory; NCI DCEG Cancer Sequencing Working Group, Maillard I., Alter B.P., Keegan C.E., Nandakumar J., and Savage S.A.2014. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev.28(19):2090–2102.
Lamm N., Ordan E., Shponkin R., Richeler C., Aker M., and Tzfati Y.2009. Diminished telomeric 3′ overhangs are associated with telomere dysfunction in Hoyeraal-Hreidarsson syndrome. PLoS ONE. 4(5):e5666.
Le Guen T., Jullien L., Touzot F., Schertzer M., Gaillard L., Perderiset M., Carpentier W., Nitschke P., Picard C., Couillault G., Soulier J., Fischer A., Callebaut I., Jabado N., Londono-Vallejo A., de Villartay J.P., and Revy P.2013. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet.22(16):3239–3249.
Malbora B., Avci Z., and Ozbek N.2014. Aplastic anemia and Hoyeraal-Hreidarsson syndrome. Skinmed.12(2):117–118.
Marrone A., Walne A., Tamary H., Masunari Y., Kirwan M., Beswick R., Vulliamy T., and Dokal I.2007. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 110(13):4198–4205.
Ozdemir M.A., Karakukcu M., Kose M., Kumandas S., and Gumus H.2004. The longest surviving child with Hoyeraal-Hreidarsson Syndrome. Haematologica. 89(9):ECR38.
Sznajer Y., Baumann C., David A., Journel H., Lacombe D., Perel Y., Blouin P., Segura J.F., Cezard J.P., Peuchmaur M., Vulliamy T., Dokal I., and Verloes A.2003. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). Eur J Pediatr.162(12):863–867.
Touzot F., Callebaut I., Soulier J., Gaillard L., Azerrad C., Durandy A., Fischer A., de Villartay J.P., and Revy P.2010. Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Scie USA107(22):10097–10102.
Touzot F., Gaillard L., Vasquez N., Le Guen T., Bertrand Y., Bourhis J., Leblanc T., Fischer A., Soulier J., de Villartay J.P., and Revy P.2012. Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J. Allergy Clin. Immunol.129(2):473–482.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 3 • September 2016
Pages: 119 - 126
History
Received: 4 May 2016
Accepted: 13 July 2016
Accepted manuscript online: 22 July 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
NufarMarcus. 2016. Immunodeficiency and severe gastrointestinal manifestations in a patient with novel DKC1 mutations causing Hoyeraal–Hreidarsson syndrome. LymphoSign Journal.
3(3): 119-126. https://doi.org/10.14785/lymphosign-2016-0006
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Abnormal apoptosis causes colitis and enteritis in patients with Hoyeraal–Hreidarsson syndrome due to DKC1 mutations
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
