IL2RG: A series of three novel mutations with clinical manifestations
Abstract
Background: X-linked severe combined immunodeficiency (SCID) is caused by mutations in the IL2RG gene and classically presents with absent T cells and natural killer (NK) cells. Mutational analysis has contributed to the understanding of this gene.
Methods: The primary immunodeficiency (PID) registry was reviewed for patients with SCID with novel IL2RG mutations. The clinical phenotype was assessed using a retrospective chart review.
Results: We describe 3 novel mutations in the IL2RG. The first was a guanine to adenine substitution at position 215 (c.215 G > A) in exon 2 leading to a cysteine to tyrosine substitution at position 72 (p.Cys72Tyr), with a typical T− B+ NK− phenotype. The second was a deletion of thymine at position 618 and adenine at position 619 (c.618_619TA) in exon 5, leading to a frameshift at the 206 amino acid (p.His206fs). The phenotype was characterized by a classic SCID presentation and immunophenotyping revealed a low number of absolute lymphocytes with mostly B cells, low levels of immunoglobulins, as well as very low NK cells. Finally, the third mutation was a guanine to cytosine substitution at position 341 (c.341 G > C) in exon 3 leading to a glycine to alanine substitution at position 114 (p.Gly114Ala), presenting with a T+ B+ NK+ phenotype. The presence of T and NK cells in IL2RG are discussed in the context of other mutations allowing for T and NK cells in IL2RG mutations, as well as in the setting of maternal engraftment.
Conclusion: To the best of our knowledge we describe 3 novel mutations in the IL2RG gene and the associated phenotypes. These mutations illustrate that these patients can have atypical immunological evaluations.
Statement of novelty: To the best of our knowledge, this paper describes 3 novel mutations in the IL2RG gene.
Introduction
X-linked severe combined immunodeficiency (SCID) is the most common form of SCID. It is caused by mutations in the IL2RG gene, and involves both the humoral and cell-mediated immune system, with typical absence of both T cells and natural killer (NK) cells. With respect to its clinical presentation, X-linked SCID typically presents in early infancy with failure to thrive, chronic diarrhea, opportunistic infections, and fatality in the first 2 years of life without immune reconstitution (Buckley 2004; van der Burg and Gennery 2011). The average incidence of SCID has been quoted at 1 in 66,250 (Kwan et al. 2013). In 2013, Ontario began screening for SCID as part of the routine newborn screening with the goal of improving early diagnosis and intervention (Ontario Ministry of Health and Long-Term Care 2013).
The IL2RG gene encodes the γc portion of the IL-2 receptor, which along with IL-2Rα and IL-2Rβ makes up the entire IL-2 receptor. The γc portion interacts with the IL-2 cytokine, and it has fundamental function in the development of regulatory T cells, maintaining peripheral tolerance, as well as increasing the cytolytic activity of NK cells (reviewed in Kovanen and Leonard (2004)). The γc portion is also involved in the signaling of various other cytokines including IL-4, IL-7 (Kawahara et al. 1994), IL-9 (Russell et al. 1994), IL-15 (Giri et al. 1994), and IL-21 (Ozaki et al. 2002). IL-7 has been shown to be important for lymphocyte homeostasis and B-cell development, whereas IL-15 is fundamental in the development of NK cells, as mice deficient in IL-15 lack both NK cells and CD8+ T cells (Kennedy et al. 2000; Lodolce et al. 1998). Finally, IL-4 and IL-21 have been implicated in T-helper cell differentiation as well as the synthesis of immunoglobulins (reviewed in Kovanen and Leonard (2004)). Taken together, these findings provide evidence that γc is fundamental for the development and function of T, B, and NK cells (Kovanen and Leonard 2004).
Mutations in the IL2RG gene leading to X-linked SCID were first described in 1993 (Noguchi et al. 1993; Puck et al. 1993) and since then, there have been 200 mutations in the 8 exons of the IL2RG gene catalogued, of which the majority (119) are frameshift mutations (Kattman and Puck 2012).
An understanding of the genetic mutation as well as the corresponding phenotype is fundamental. Mutational analysis has revealed that there are parts of the gene prone to mutation; these are known as hotspots (Puck et al. 1993). In addition, genotype/phenotype correlations have been revealed as evidenced most strikingly by the R222C mutation of IL2RG mutation (Fuchs et al. 2014; Sharfe et al. 1997). The R222C mutation of IL2RG leading to a SCID phenotype presented atypically with a normal thymus gland, mitogen response, as well as normal numbers of T and B cells. However, despite having a normal number and repertoire of T cells, the T cells were found to have a reduced ability to bind to IL-2. Interestingly, the index case presented with P. jiroveci pneumonia at 9 months of age, in keeping with an underlying immunodeficiency (Sharfe et al. 1997). This finding provides evidence that different genetic mutations in the IL2RG can lead to variable SCID phenotypes. In this study, we sought to further the knowledge of specific mutations in the IL2RG, through a description of 3 novel mutations in the IL2RG gene and the associated phenotype.
Methods
Patients
The primary immunodeficiency (PID) registry was reviewed for patients with SCID with novel IL2RG mutations. The registry and analysis of SCID patients is approved by the institutional research ethics board. Genetic testing was performed with informed consent.
Lymphocyte enumeration and immunoglobulin determinations were performed in a clinical laboratory.
Genetic analysis
An automated sequencer (Beckman Coulter CEQ8000) was used to generate sequencing of all exons and exon–intron boundaries of IL2RG. These data were compared with the wild-type genetic sequence. Novelty of the genetic change was assessed by search of the Leiden Open Variation Database for IL2RG (Kattman and Puck 2012) as well as NCBI databases (http://www.ncbi.nlm.nih.gov/).
Results
Clinical details
Patient 1 was diagnosed at 7.5 months of age with a T− SCID. He initially presented at 6 months of age with a cough of unknown etiology and vomiting. In terms of infectious history, he had oral thrush, extensive bilateral pneumonia, chronic diarrhea that was positive for C. difficile, as well as a urinary tract infection positive for E. coli. Patient 1 also presented with failure to thrive with a height in the 25th percentile and weight less than the third percentile. He also had normocytic anemia (Table 1).
Table 1:
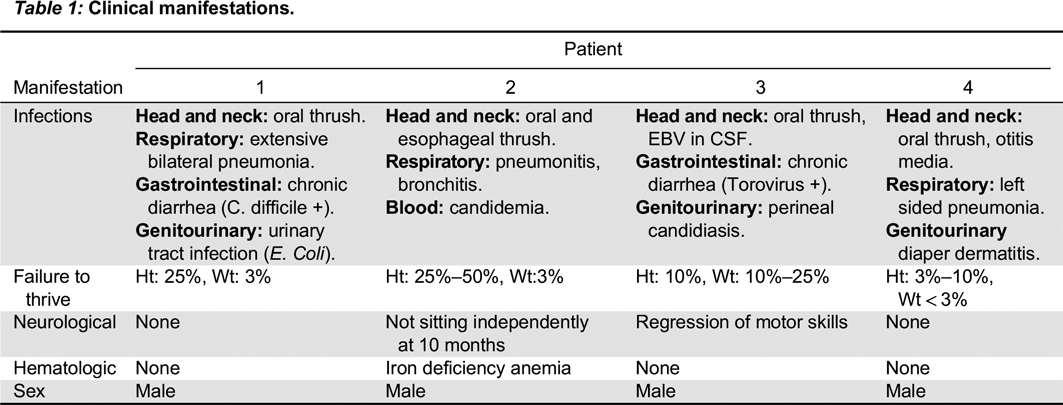
Patient 2 was diagnosed at 10 months of age with T− SCID. He initially presented at 6 months of age with oral and esophageal thrush resistant to medical therapy. At 7 months of age, he was diagnosed with pneumonitis and bronchitis and was subsequently found to have Parainfluenza. At 10 months of age, he was admitted for chronic diarrhea, vomiting, feeding intolerance, and failure to thrive. He had oral candidiasis, esophagitis, as well as candidemia. Developmentally, he was not yet sitting independently. Anthropometric analysis revealed a height between the 25th and 50th percentiles and weight of 7.1 kg, which corresponds to the third percentile (Table 1).
Patient 3, the cousin of Patient 2, was diagnosed at 3 months of life with T− SCID. Patient 3 presented with diarrhea and CNS involvement with both regression in motor skills, as well as the presence of EBV in the CSF. Patient 3 had oral thrush, perineal candidiasis, as well as chronic diarrhea that was positive for torovirus. He had failure to thrive with a height in the tenth percentile and a weight between the 10th and 25th percentile (Table 1).
Patient 4 was diagnosed at 7 months of age with a T+ combined immunodeficiency He initially began having symptoms at 4 months of age and was found to have oral thrush, diaper dermatitis, left-sided pneumonia, as well as otitis media and nasal discharge. There was no history of CNS involvement or gastrointestinal involvement. He had failure to thrive, and at 7 months was found to have a height between the third and tenth percentiles and a weight less than the third percentile (Table 1).
Immunophenotype
The immune investigations are detailed in Table 2. An abnormal immunophenotype was characterized. Patient 1 had evidence of absent T cells, a high number of B cells, and absent NK cells. Patient 1 had an absolute lymphocyte count of 1080 cells/µL with a total of 7 CD3+ cells/µL and undetectable CD4+ or CD8+. The levels of CD56+ cells were undetectable. Phytohaemagglutinin (PHA) stimulation was low. Of the total number of lymphocytes, 1069 cells/µL were B cells (99%), with trace IgG and no detectable IgM or IgA (Table 2).
Table 2:
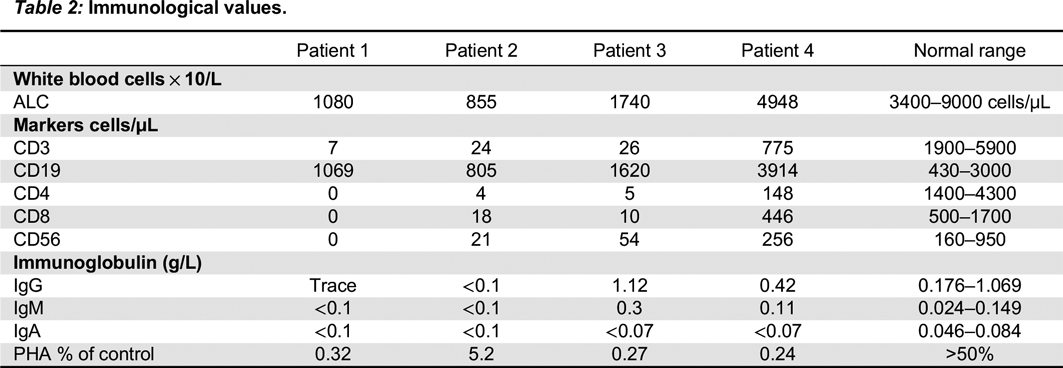
Patient 2 had T-cell lymphopenia, with an absolute lymphocyte count of 855 cells/µL, with 4 cells/µL CD4 cells (0.4%) and 18 cells/µL CD8 cells (2.2%). The total CD3+ cells were 24 cells/µL. There were a high number of B-lymphocytes with 805 cells/µL CD19+ cells, which corresponded to 94% of the cells and a small number of circulating NK cells with a total number of 21 cells/µL CD56+ cells (2.4 %). IgG, IgM, and IgA levels were all less than 0.1 g/L. Finally, the PHA stimulation index was low (Table 2).
For patient 3, an abnormal immunophenotype was characterized with evidence of T-cell lymphopenia, a high number of B cells, and few NK cells. Patient 3 had an absolute lymphocyte count of 1740 cells/µL with 5 cells/µL CD4+ cells (0.3%) and 10 cells/µL CD8+ T cells (0.6%). Patient 3 had a total of 26 CD3+ cells/µL cells. With respect to a high number of B cells, patient 3 had 1620 (93%) CD19+ cells/µL. The immunoglobulin levels were 1.12 g/L IgG, 0.3 g/L IgM, and <0.07 g/L IgA. The PHA stimulation was low. Finally, patient 3 had a low number of NK cells with 54 cells/µL (3.1%) CD56+ cells (Table 2).
For patient 4, an abnormal immunophenotype was also characterized including T-cell lymphopenia, high levels of B cells, and the presence of NK cells. Patient 4 had an absolute lymphocyte count of 4948 cells/µL with 775 cells/µL CD3+ cells, 148 cells/µL CD4+ cells (3.0%) and 446 cells/µL CD8+ T cells (9.0%). There were 3914 CD19+ cells/µL (79%). The immunoglobulins were IgG level of 0.42 g/L, IgM of 0.11 g/L, and IgA of <0.7 g/L. There were 256 cells/µL CD56+ cells indicating a normal number of circulating NK cells. Finally, the PHA stimulation was low. Karyotype could not be performed due to mitogen failure. HLA typing found both maternal haplotypes and one paternal haplotype (Table 2).
Genetic analysis
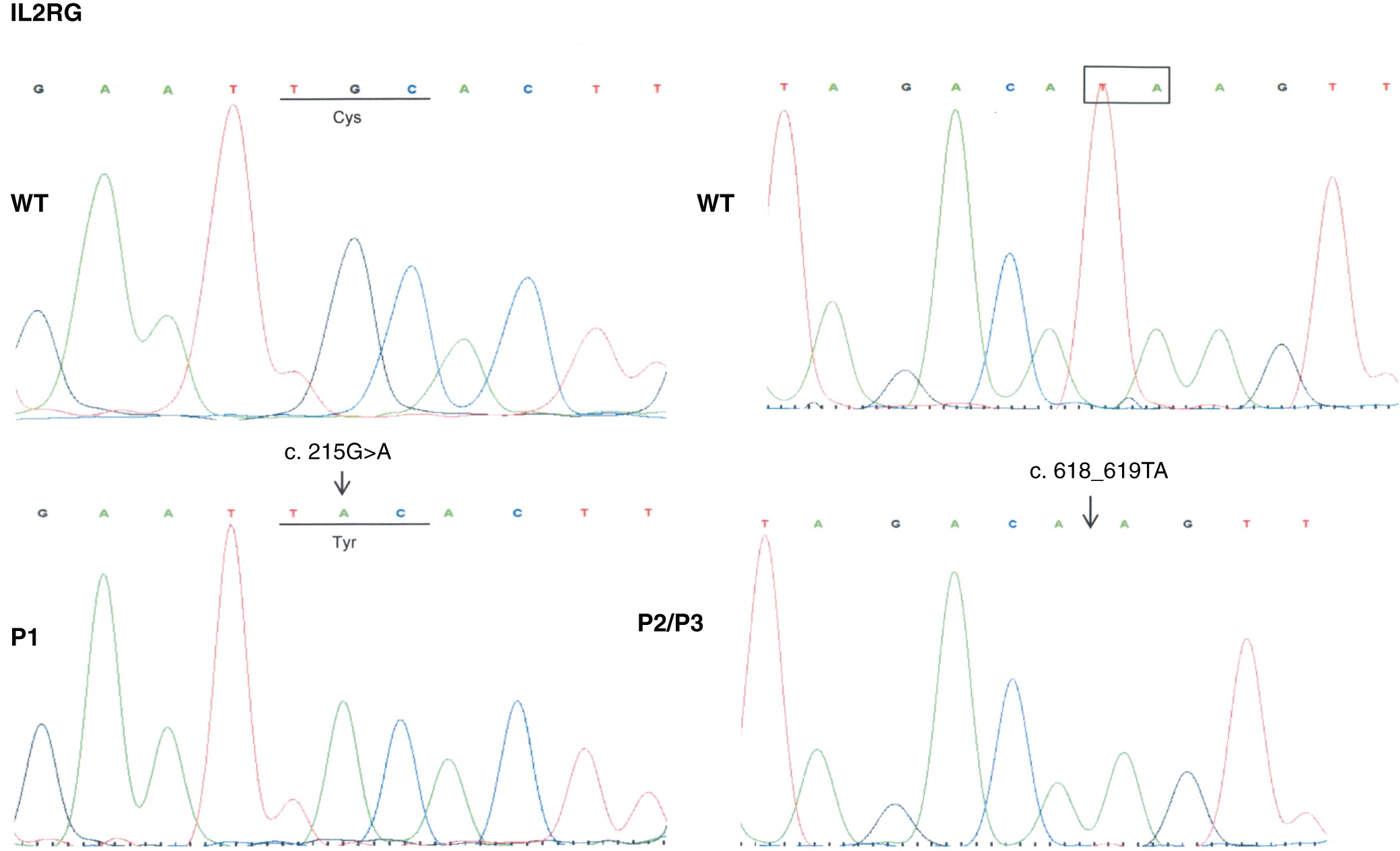
Patient 1 had a guanine to adenine substitution at position 215 (c.215 G > A) in exon 2, which predicts a cysteine to tyrosine substitution at position 72 (p.Cys72Tyr) in the IL-6 receptor alpha binding domain (Table 3).
Table 3:
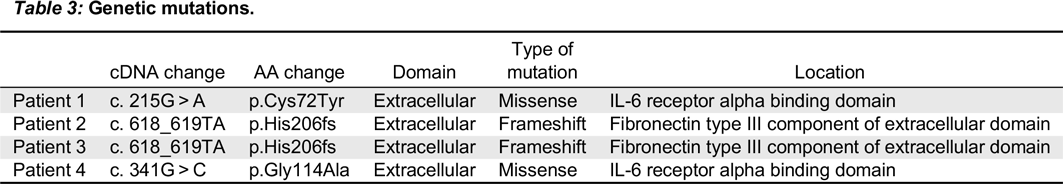
Patients 2 and 3 had a deletion of a tyrosine at position 618 and adenine at position 619 in exon 5 (c. 618_619TA), which predicts a deletion of a histidine amino acid at position 206 (p.His206fs) in the fibronectin type III component of the extracellular domain (Table 3).
Patient 4 had a guanine to cytosine substitution at position 341 (c. 341 G > C) in exon 3, which predicts a glycine to alanine substitution at position 114 (p.Gly114Ala) in the IL6 receptor alpha-binding domain (Table 3).
The mutation locations with respect to the various domains of the IL2RG gene are shown in Figure 1. The electropherogram diagrams are shown in Figure 2; however, the data for Patient 4 are unavailable.
Figure 1:
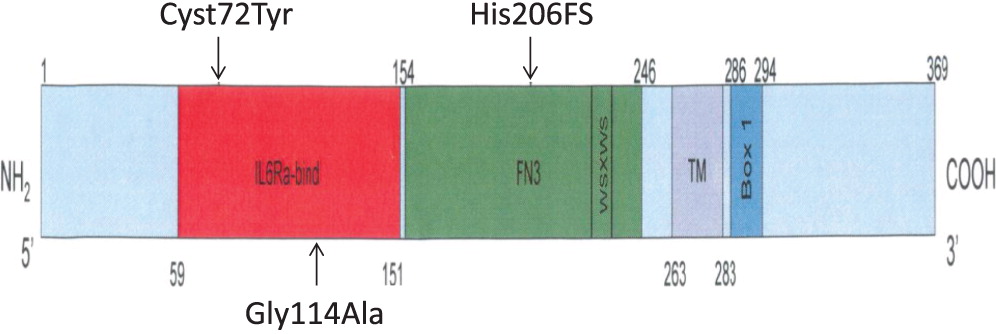
Figure 2:
Discussion
We describe 3 novel mutations in the IL2RG gene leading to SCID phenotypes.
In the first mutation, Patient 1 had a c. 215 G > A substitution, which predicts a p.Cys72Tyr in the extracellular domain. This corresponded to a phenotype of T− B+ NK−, in keeping with a typical X-linked SCID phenotype. Typically, mutations in IL2RG lead to a T− NK− phenotype, as the development and growth of both T and NK cells depend on various cytokines including IL-2, IL-4, IL-7, IL-15, and IL-21 all of which signal through the γc receptor (Giri et al. 1994; Kawahara et al. 1994; Russell et al. 1994; Ozaki et al. 2002). A lack of NK cells in mutations in the γc receptor are thought to be secondary to impaired IL-15 signaling, as mice deficient in the IL-15 receptor alpha subunit have a significant block in NK-cell development (Lodolce et al. 1998; Notarangelo et al. 2000).
In the second mutation, a c. 618_619TA in exon 5 lead to a p.His206fs deletion. Both patients had similar clinical presentations. Specifically, both Patients 2 and 3 were noted to have classic SCID presentations of fungal infections with candidiasis, viral infections, chronic diarrhea, as well as failure to thrive. With respect to their laboratory findings, both patients had a low number of absolute lymphocytes, T cells, and their lymphocytes were comprised of mostly B cells. Taken together, these data provide evidence that a frameshift at position 206 leads also to a typical IL2RG SCID phenotype. This provides evidence of a strong genotype and phenotype correlation between Patients 2 and 3.
In the last mutation, Patient 4 had c. 341 G > C in exon 3, which predicts a p.Gly114Ala substitution in the extracellular domain. The phenotype was characterized as a T+ B− NK+. We note that he did have evidence of some maternal engraftment. Interestingly, Puck et al. (1993) described a different missense mutation at this site with a p.Gly114Ala mutation (cDNA position 355 exon 3). The patient did produce the IL2RG mRNA which, supports that this mutational site could possibly lead to a presentation of leaky SCID. There are a variety of causes of T+ SCID including Omenn’s syndrome (reviewed in (Villa et al. 2008) and maternal engraftment. Some degree of maternal engraftment is common in SCID. In a series of 121 SCID patients, some maternally engrafted cells were detected in 48 patients (Muller et al. 2001). Furthermore, a late presentation of X-linked SCID was described secondary to revertant somatic mosaicism (Stephan et al. 1996), and more recently, this phenomenon has been described with a mutation in the P58T region of the IL2RG (Okuno et al. 2015). In our patient, it is not possible to say which proportions of his cells were of maternal origin versus possibly a leaky phenotype. Therefore we will explore some other examples of T+ SCID.
T+ SCID has been described in other cases of IL2RG mutations. The R222C mutation, is a notable example of this phenomenon (Sharfe et al. 1997). As previously mentioned, the patient presented with pneumocystis jiroveci pneumonia at 9 months of age and had normal T-cell, B-cell, and NK-cell numbers, a complete T-cell repertoire and normal response to mitogenic stimulation. However, the patient did not respond to exogenous IL-2, despite having normal expression levels of the IL2RG protein. The patient was found to have an altered IL-2 binding site leading to a reduced affinity for IL-2 binding, and subsequent downstream activation of the Janus kinase 3 pathway (Sharfe et al. 1997). A follow-up report described more patients with this mutation, and stressed that IL2RG R222C mutation needs to be ruled out in children with clinical features of SCID, even in the absence of typical laboratory data (Somech and Roifman 2005). Further studies on the R222C mutation confirmed the findings of the functional impairment in T-cell response to IL-2. They also found that for NK cells, IL-2 mediated NK-cell degranulation, as well as IL-15 induced cytokine expression was significantly reduced (Fuchs et al. 2014). Recently, a different mutation in the IL2RG was described leading to a similar T+ SCID phenotype. A 12-base pair intronic deletion, c609-15_609-4del12 was described in a T+ B− NK− X-linked SCID patient (Gray et al. 2015). The patient had mild T-cell lymphopenia, normal T-cell receptor diversity, a reduced PHA stimulation, and no evidence of chimerism. This deletion lead to a splicing defect and a subsequent truncated IL2RG mRNA, with normal IL-2Rγ expression. There was also complete absence of IL-2 and IL-7 mediated STAT5 phosphorylation (Gray et al. 2015). Taken together, these findings provide evidence that mutations in the IL2RG can lead to a T+ SCID with normal or mildly reduced numbers of T cells, but impairment in IL-2 mediated downstream signaling cascades that are vital to T-cell function.
The presence of normal numbers of NK cells in patient 4 deserves discussion. NK cells of maternal origin have been reported in IL2RG deficiency. Recently, Estevez et al. (2014) described a case of an 8-month-old boy who presented with diarrhea and dehydration and was found to have S. auricularis, S. epidermidis, and C. parasilopsis in the blood cultures, with stool cultures positive for both C. albicans and P. aeruginosa. The patient was subsequently found to have an ACC insertion in exon 5, of IL2RG, with a phenotype of T− B+ NK+, and he was shown to have maternally derived NK cells. Normal numbers of NK cells have been seen in some previous IL2RG mutations. Enumeration of NK cells was normal in all reports of R222C (Fuchs et al. 2014). Additionally, there have been a few reports of the presence of NK cells in patients with mutations involving the γc portion of the IL2RG and these have raised the possibility of a potential downstream activation mechanism in NK cell differentiation. In one case report, a patient with a mutation in exon 5 with a c.691G > A corresponding to a R226H substitution, had increased NK cells (59.5%) with an erythroderma-like skin reactive characterized by infiltration of CD56+ cells. These were not maternal in origin as evidenced by the microsatellite polymorphism analysis where the allelic patterns from the patient were distinct from maternal ones (Shibata et al. 2007). A second case report described a mutation in a splice site region in intron 3, leading to 2 alternatively spliced mRNAs and undetectable γc levels. The patient, however, had near normal numbers of NK cells with a genotype of T− B+ NK+. From this study, the authors concluded that the IL-15 receptor-mediated signaling is preferentially retained when γc levels are depleted (Ginn et al. 2004). Taken together, these reports suggest that NK cells in IL2RG deficiency may be of maternal origin, but also that there may be downstream pathways for the activation and differentiation of NK cells in γc deficiency, and that not all detectable NK cells in the IL2RG deficiency are of maternal origin. Further studies will need to further characterize the mutations and their implications on T and NK cells.
In conclusion, we described 3 novel mutations in the IL2RG gene leading to various phenotypic presentations of X-linked SCID. We identified a mutation with a p.Cys72Tyr substitution, which was in keeping with the typical phenotype findings of X-linked SCID. In the second mutation, a deletion of p.His206fs, both Patient B and C were found to have SCID presentations with very low T and NK cells. Finally, we described a novel missense mutation leading to a p.Gly114Ala substitution, which presented as a T+ NK+ SCID. This finding further strengthens the knowledge that patients with IL2RG mutations can have atypical presentations. These descriptions of 3 novel mutations contribute to the molecular understanding of IL2RG. Future studies will need to address the p.Gly114Ala mutation and its effect on IL-2 mediated downstream signaling cascades.
REFERENCES
Buckley R.H.2004. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol.22:625–655.
Estevez O.A., Ortega C., Fernandez S., Aguado R., Rumbao J., Perez-Navero J., and Santamaria M.2014. A novel IL2RG mutation presenting with atypical T(-)B(+)NK+ phenotype: rapid elucidation of NK cell origin. Pediatr. Blood Cancer.61(1):178–179.
Fuchs S., Rensing-Ehl A., Erlacher M., Vraetz T., Hartjes L., Janda A., Rizzi M., Lorenz M.R., Gilmour K., de Saint-Basile G., Roifman C.M., Cheuk S., Gennery A., Thrasher A.J., Fuchs I., Schwarz K., Speckmann C., and Ehl S.2014. Patients with T(+)/low NK(+) IL-2 receptor gamma chain deficiency have differentially-impaired cytokine signaling resulting in severe combined immunodeficiency. Eur. J. Immunol.44(10):3129–3140.
Ginn S.L., Smyth C., Wong M., Bennetts B., Rowe P.B., and Alexander I.E.2004. A novel splice-site mutation in the common gamma chain (gammac) gene IL2RG results in X-linked severe combined immunodeficiency with an atypical NK+ phenotype. Hum. Mutat.23(5):522–523.
Giri J.G., Ahdieh M., Eisenman J., Shanebeck K., Grabstein K., Kumaki S., Namen A., Park L.S., Cosman D., and Anderson D.1994. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J.13(12):2822–2830.
Gray P.E., Logan G.J., Alexander I.E., Poulton S., Roscioli T., and Ziegler J.2015. A novel intronic splice site deletion of the IL-2 receptor common gamma chain results in expression of a dysfunctional protein and T-cell-positive X-linked Severe combined immunodeficiency. Int. J. Immunogenet.42(1):11–14.
Kattman B. and Puck J.2012. LOVD at NCBI. interleukin 2 receptor, gamma (IL2RG). Available at: http://www.ncbi.nlm.nih.gov/lovd/home.php?select_db=IL2RG.
Kawahara A., Minami Y., and Taniguchi T.1994. Evidence for a critical role for the cytoplasmic region of the interleukin 2 (IL-2) receptor gamma chain in IL-2, IL-4, and IL-7 signalling. Mol. Cell. Biol.14(8):5433–5440.
Kennedy M.K., Glaccum M., Brown S.N., Butz E.A., Viney J.L., Embers M., Matsuki N., Charrier K., Sedger L., Willis C.R., Brasel K., Morrissey P.J., Stocking K., Schuh J.C., Joyce S., and Peschon J.J.2000. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med.191(5):771–780.
Kovanen P.E. and Leonard W.J.2004. Cytokines and immunodeficiency diseases: critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev.202:67–83.
Kwan A., Church J.A., Cowan M.J., Agarwal R., Kapoor N., Kohn D.B., Lewis D.B., McGhee S.A., Moore T.B., Stiehm E.R., Porteus M., Aznar C.P., Currier R., Lorey F., and Puck J.M.2013. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J. Allergy. Clin. Immunol.132(1):140–150.e7.
Lodolce J.P., Boone D.L., Chai S., Swain R.E., Dassopoulos T., Trettin S., and Ma A.1998. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity.9(5): 669–676.
Muller S.M., Ege M., Pottharst A., Schulz A.S., Schwarz K., and Friedrich W.2001. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: A study of 121 patients. Blood.98(6):1847–1851.
Noguchi M., Yi H., Rosenblatt H.M., Filipovich A.H., Adelstein S., Modi W.S., McBride O.W., and Leonard W.J.1993. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell.73(1):147–157.
Notarangelo L.D., Giliani S., Mazza C., Mella P., Savoldi G., Rodriguez-Perez C., Mazzolari E., Fiorini M., Duse M., Plebani A., Ugazio A.G., Vihinen M., Candotti F., and Schumacher R.F.2000. Of genes and phenotypes: the immunological and molecular spectrum of combined immune deficiency. Defects of the gamma(c)-JAK3 signaling pathway as a model. Immunol. Rev.178:39–48.
Okuno Y., Hoshino A., Muramatsu H., Kawashima N., Wang X., Yoshida K., Wada T., Gunji M., Toma T., Kato T., Shiraishi Y., Iwata A., Hori T., Kitoh T., Chiba K., Tanaka H., Sanada M., Takahashi Y., Nonoyama S., Ito M., Miyano S., Ogawa S., Kojima S., and Kanegane H.2015. Late-Onset Combined Immunodeficiency with a Novel IL2RG Mutation and Probable Revertant Somatic Mosaicism. J. Clin. Immunol.35(7):610–614.
Ontario Ministry of Health and Long-Term Care. 2013. Newborn Screening Ontario to Include Severe Combined Immune Deficiency. Available at: https://news.ontario.ca/mohltc/en/2013/08/newborn-screening-ontario-to-include-severe-combined-immune-deficiency.html.
Ozaki K., Spolski R., Feng C.G., Qi C.F., Cheng J., Sher A., Morse H.C. III, Liu C., Schwartzberg P.L., and Leonard W.J.2002. A critical role for IL-21 in regulating immunoglobulin production. Science.298(5598):1630–1634.
Puck J.M., Deschenes S.M., Porter J.C., Dutra A.S., Brown C.J., Willard H.F., and Henthorn P.S.1993. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum. Mol. Genet.2(8):1099–1104.
Russell S.M., Johnston J.A., Noguchi M., Kawamura M., Bacon C.M., Friedmann M., Berg M., McVicar D.W., Witthuhn B.A., and Silvennoinen O.1994. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science.266(5187):1042–1045.
Sharfe N., Shahar M., and Roifman C.M.1997. An interleukin-2 receptor gamma chain mutation with normal thymus morphology. J. Clin. Invest.100(12):3036–3043.
Shibata F., Toma T., Wada T., Inoue M., Tone Y., Ohta K., Kasahara Y., Sano F., Kimura M., Ikeno M., Koizumi S., and Yachie A.2007. Skin infiltration of CD56(bright) CD16(-) natural killer cells in a case of X-SCID with Omenn syndrome-like manifestations. Eur. J. Haematol.79(1):81–85.
Somech R. and Roifman C.M.2005. Mutation analysis should be performed to rule out gammac deficiency in children with functional severe combined immune deficiency despite apparently normal immunologic tests. J. Pediatr.147(4):555–557.
Stephan V., Wahn V., Le Deist F., Dirksen U., Broker B., Muller-Fleckenstein I., Horneff G., Schroten H., Fischer A., and de Saint Basile G.1996. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N. Engl. J. Med.335(21):1563–1567.
van der Burg M. and Gennery A.R.2011. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur. J. Pediatr.170(5):561–571.
Villa A., Notarangelo L.D., and Roifman C.M.2008. Omenn syndrome: inflammation in leaky severe combined immunodeficiency. J. Allergy. Clin. Immunol.122(6):1082–1086.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 3 • September 2016
Pages: 111 - 118
History
Received: 21 February 2016
Accepted: 17 May 2016
Accepted manuscript online: 19 May 2016
Version of record online: 19 May 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AlishaJamal and Julia E.M.Upton. 2016. IL2RG: A series of three novel mutations with clinical manifestations. LymphoSign Journal.
3(3): 111-118. https://doi.org/10.14785/lymphosign-2016-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
