Introduction
Purified immunoglobulin (IgG) has been used for more than half a century as a therapeutic modality to replace antibody deficiencies. Early immunoglobulin preparations were restricted to intramuscular injection (
Janeway and Rosen 1966), and were clearly sub-optimal, as trough levels were low and insufficient for prevention of major infections (
Eibl 2008). The introduction of IgG preparations suitable for intravenous use (IVIG), which were free from trace contaminants, revolutionized the treatment of these conditions (
Nolte et al. 1979;
Hooper 2008).
Roifman et al (1987) have demonstrated that in patients with agammaglobulinemia, maintaining an IgG serum level of more than 500 mg/dL was superior to lower concentrations in preventing serious infections, as well as in improving lung disease. This threshold has been used for decades by clinicians as a minimum required trough level in patients with minimal endogenous IgG (
Roifman et al. 1987;
Orange et al. 2006;
Eibl 2008). Subsequently, several reports have demonstrated that higher trough levels (700–900 mg/dL) may have further benefits in protecting patients from all types of infections (
Roifman et al. 1987;
Eijkhout et al. 2001;
Orange et al. 2010).
Modern IVIG products have an excellent safety record and incorporate multiple effective measures of virus inactivation or removal in their manufacture (
Hooper 2008). However, tolerability of products appears to be variable, with some patients tolerating some products but not others. It is therefore important to introduce new IVIG products to provide additional options for patients. Introduction of new safe products is also needed to generate a steady and adequate supply of a limited resource.
Kedrion IVIG 10% (Human) is a modern 10% liquid IVIG preparation produced using Cohn–Oncley fractionation (
Cohn et al. 1946;
Oncley et al. 1949) and anion-exchange chromatography. Several measures are employed to achieve virus inactivation/removal, including treatment with solvent detergent and nanofiltration.
We report here the results of a prospective multicenter clinical trial that studied the efficacy, pharmacokinetics (PK), safety, and tolerability of Kedrion IVIG 10% in patients with primary immunodeficiency (PID).
Methods
Study product
Kedrion IVIG 10% is a 10% (weight/volume) IgG (Human) solution for intravenous (IV) injection derived from US plasma donors and produced by Kedrion, S.p.A. at their manufacturing facilities in Bolognana (Lucca), Italy.
The drug product contains approximately 100 mg/mL of human plasma protein, at least 96% of which is IgG. The majority of the IgG protein (>90%) is present in a monomer plus dimer form. IgG molecules may interact forming aggregates and polymers. The product specification limits for polymers and aggregates is ≤3%. The maximum IgA content is 50 μg/mL. Other excipients include glycine (functioning as a stabilizer), sodium chloride, and water for injection. The product contains no preservatives.
Kedrion IVIG 10% has a pH between 4.0 and 5.5, with a mean osmolality of 287 mOsmol/kg (range: 267–296 mOsmol/kg; n = 15 clinical lots).
The Kedrion 10% Immune Globulin (Human) manufacturing process consists of the following steps that have been validated for their ability either to remove or inactivate viral pathogens:
•
Cold ethanol fractionation:
◦
First step – precipitation of fraction I
◦
Second step – precipitation and filtration of fraction II+III
•
Solvent/detergent treatment
Kedrion introduced an additional purification step (the first is the II+III precipitation), with sodium caprylate precipitation, whose efficacy in precipitation of contaminants and impurities is well known (
Parkkinen et al. 2006). Sodium caprylate precipitates α and β globulins, lipids, kallikrein, and other contaminants. The precipitation with sodium caprylate has been shown to remove procoagulant activity and reduces the content of Coagulation Factor XIa by more than 99% (evaluated by thrombin generation assay).
After sodium caprylate contact, the solution is filtered and concentrated. It then undergoes solvent/detergent treatment. The solution is further purified by ion-exchange chromatography using a strong anion exchange resin that binds IgG. The recovered IgG solution is then diafiltered with a solution of saline and glycine and the pH is lowered to 4.0–4.5. It is then subjected to a specific step of viral removal, which is nanofiltration through a 20 nm filter. The nanofiltered solution is finally concentrated to the target protein concentration of 90–110 g/L. The final formulated IgG is bulk sterile filtered and aseptically filled into sterile, depyrogenated glass vials. Lastly, the final product undergoes low-pH incubation (a specific step of viral inactivation) and quarantine.
Kedrion IVIG 10% is derived from a minimum of 1000 plasma donors, thus, is expected to contain a broad spectrum of antibody specificities.
Study design
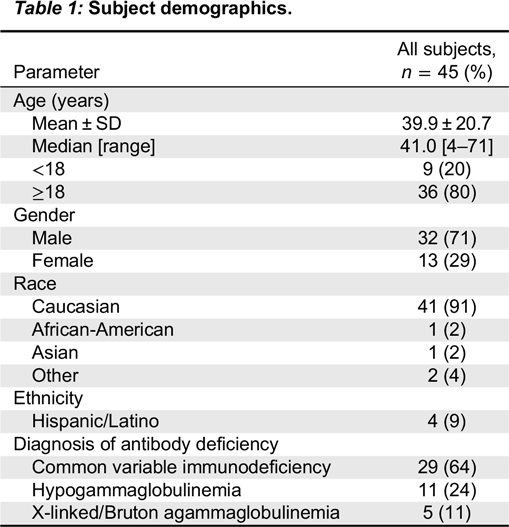
This was a prospective, open-label, single-arm, multi-centre, historically controlled, Phase III study measuring the efficacy, PK, safety, and tolerability of Kedrion 10% IVIG in patients with hypogammaglobulinemia and antibody deficiency. Prior to study enrollment, the diagnosis of PID was confirmed and included a review of infection history, serum IgG concentration prior to IgG therapy, and antibody responses to vaccination with polysaccharide and protein vaccines. The study design required at least 40 evaluable subjects aged 2–70 years, with well-defined PID and documented agammaglobulinemia or hypogammaglobulinemia, to be treated for 12 months to determine the rate of serious bacterial infections, and up to 30 patients were to be assessed for PK of the study product.
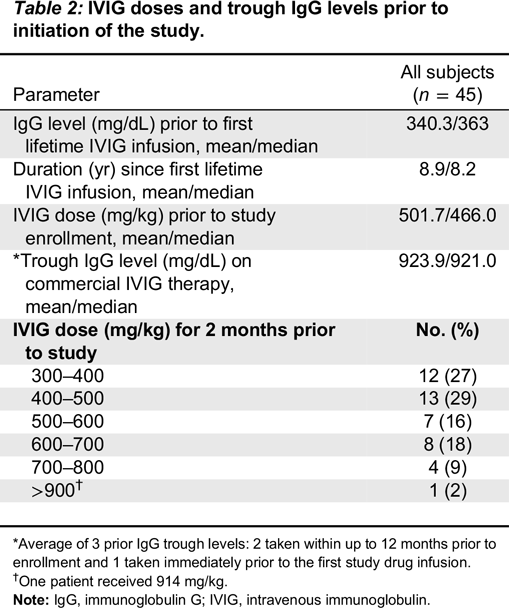
Upon consenting and screening, eligible subjects received IV infusions of the study product at the same dose and interval as used for their previous IVIG maintenance therapy. Kedrion IVIG 10% was administered at a dose of 300–900 mg/kg (of body weight) every 21 or 28 days (±4 days; depending on their pre-study IVIG treatment schedule) for a period of 12 months. The dose regimen remained unchanged throughout the study period unless there was a medically justified need to modify it. The total subject follow-up was approximately 14 months, during which subjects underwent 12 months of treatment, 13 treatment visits (subjects on 28-day infusion schedule) or 17 treatment visits (subjects on 21-day infusion schedule), and a follow-up visit 3 or 4 weeks after the last study drug infusion.
During the treatment and follow-up periods, patients underwent the following evaluations: adverse event monitoring and recording of concomitant medications, review of subject diaries, physical exam (prior to each infusion and at the follow-up visit), multiple vital signs assessments during the infusion and immediately after it was completed, and routine laboratory safety tests (hematology, blood chemistry, and urinalysis completed before each infusion and at the follow-up visit). Measurements of direct anti-globulin (DAT/Coombs) were performed before and after the first infusion, before and 24 hours after the fifth infusion, and at the follow-up visit. Viral safety testing was performed several times during the study and at its conclusion. PK assessments on 31 subjects were undertaken before and after the fifth study infusion.
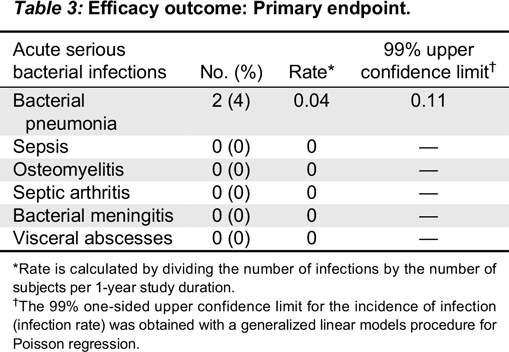
The predefined primary efficacy endpoint was the incidence of acute serious bacterial infections (defined by the US Food and Drug Administration (FDA) as bacterial pneumonia, bacteremia/sepsis, osteomyelitis/septic arthritis, bacterial meningitis or visceral abscess) per subject during the 12-month treatment period (
US FDA 2008). Serious adverse events (SAEs) potentially qualifying as primary endpoint events were adjudicated by the study Data and Safety Monitoring Board. Formal statistical analysis (noninferiority test) was applied for the occurrence of acute serious bacterial infection per subject. An upper 99% one-sided confidence limit of <1 per subject per year for the incidence of acute serious bacterial infection was predefined as an indicator of acceptable efficacy. To estimate the annualized event rate and develop the appropriate one-sided 99% upper confidence limit, a generalized linear models SAS procedure for Poisson regression PROC GENMOD was used. This procedure involves estimating a scale parameter of over-dispersion while it permits investigation of covariates, and it also allows for differential follow-up of subjects.
Summary of the inclusion criteria
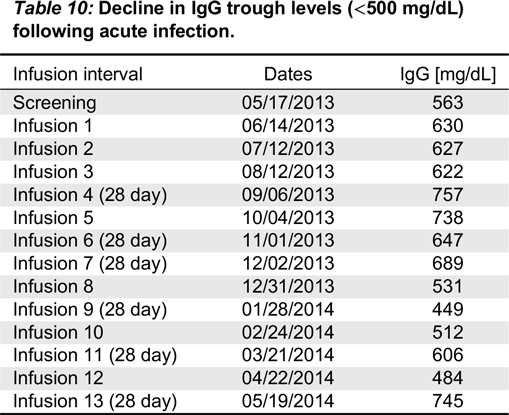
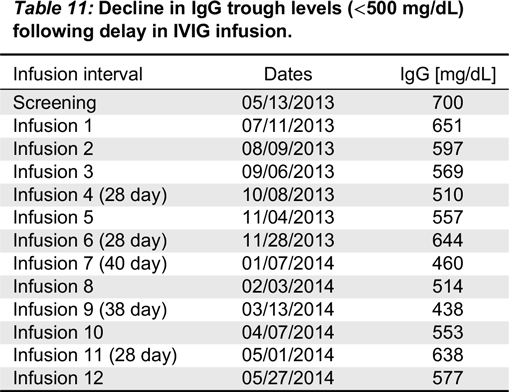
Eligible patients were males or females aged 2–70 years, with PID who required treatment with IVIG. In addition to documented agammaglobulinemia or hypogammaglobulinemia, documented antibody deficiency was required. Subjects had to have received 300–900 mg/kg of a licensed IVIG therapy at 21- or 28-day intervals for at least 3 treatment cycles prior to study enrollment; if receiving subcutaneous immunoglobulin (SCIG), subjects had to switch to IGIV for 3 infusion cycles to establish a steady state prior to receiving Kedrion IVIG 10%. All subjects had to have at least 2 documented IgG trough levels of ≥ 500 mg/dL, obtained at 2 scheduled infusions (21 or 28 days) within 12 months (including one within 6 months) prior to study enrollment. Other immunoglobulin products were prohibited for 1 infusion cycle (21 or 28 days) prior to the first infusion of Kedrion 10% IVIG and until the completion of the first follow-up visit.
To participate in the study, all subjects or parents/guardians (in case of pediatric subjects, defined as 2–17 years of age at study entry) provided written informed consent. In addition, pediatric subjects completed assent forms as appropriate per local regulations.
Subjects receiving blood products other than human albumin or human immunoglobulin within 12 months prior to enrollment and subjects who had participated in another clinical study within 3 weeks prior to enrollment were excluded. The use of corticosteroids (oral or parenteral daily dose of ≥0.15 mg/kg/day of prednisone or equivalent), other immunosuppressive drugs or chemotherapy were prohibited.
Summary of the main exclusion criteria
Patients were excluded from the study if they had any of the following: secondary immunodeficiency; newly diagnosed, previously untreated PID; dysgammaglobulinemia; isolated IgG subclass deficiency; history of repeated reactions or hypersensitivity to IVIG or other injectable forms of IgG; IgA deficiency with known antibodies to IgA; lifetime history of any thrombotic event; significant protein-losing enteropathy, nephrotic syndrome, or lymphangiectasia; acute infection within 7 days prior to screening; known history of, or having tested positive at enrollment for, human immunodeficiency virus (HIV) type 1, HIV type 2, hepatitis B virus, hepatitis C virus, hepatitis A virus, or parvovirus B19; history of epilepsy or severe migraines, or transient ischemic attack; profound anemia or persistent severe neutropenia or lymphopenia; liver function tests (transaminases) above 2.5 times the upper limit of normal; presence of a severe chronic disease or other condition precluding safe participation in the study. Other gender-specific exclusion criteria included current or planned pregnancy, breastfeeding, or refusal to use adequate contraception for women of childbearing potential.
Evaluation of PK
PK evaluation was performed at or after infusion 5 of the study to wash out residual previous IVIG or SCIG products. Blood samples were taken before the infusion and at: 30 minutes, 2 hours, 24 hours, 3 (± 1) days, 7 (± 1) days, 14 (± 1) days, and either 21 (± 1) days or 28 (± 1) days following infusion. Samples were tested for the concentration of total IgG, IgG subclasses and specific antibody titers against tetanus toxoid, Streptococcus pneumoniae, Hemophilus influenzae type b, and measles. PK calculations related to total IgG and specific IgG antibodies included the following: maximum serum concentration (Cmax), minimum serum concentration (Cmin), the time to reach maximum concentration (Tmax), IgG elimination half-life (T1/2), the area under the concentration-time curve over one dosing interval (AUC0-t), the area under the concentration-time curve up to the last measurable concentration (AUC0-tau), the area under the concentration-time curve to infinity (AUC0-inf), and terminal elimination rate constant (λz). In addition, for total IgG, the following were analyzed: total body clearance (CL), volume of distribution at steady state (weight adjusted) (Vss), terminal exponential volume of distribution (weight adjusted) (Vz), incremental recovery (IR), and mean residence time (MRT). The degree of fit (R2) of the slope of the regression line had to be ≥0.80 for the reporting of t1/2, λz, and AUC0-inf, Vss, Vz, and MRT.
Discussion
This clinical trial was conducted to evaluate the efficacy, safety, tolerability, and bioavailability of Kedrion 10% IVIG in patients with primary immunodeficiency. Current FDA and European Medicines Agency guidelines for IVIG studies were followed (
US FDA 2008;
European Medical Agency 2010), except for the pediatric enrollment being lower than required by both regulatory agencies for market approval.
Kedrion 10% IVIG was well tolerated by patients, as the rate of treatment-related AEs was low and the majority of adverse reactions were mild. Direct Coombs test was positive transiently in 47% of subjects, but it had no clinical significance, as observed in other clinical trials of IVIG (
Roifman et al. 2003). Importantly, no evidence of hemolysis was recorded during the study period in any subject.
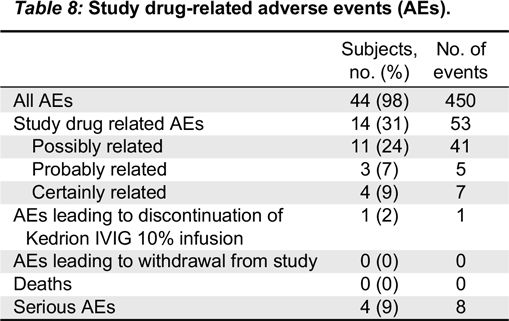
No deaths occurred during the study period, and the 8 SAEs that occurred in 4 subjects during the study period were all clearly assessed as unrelated to the study treatment product.
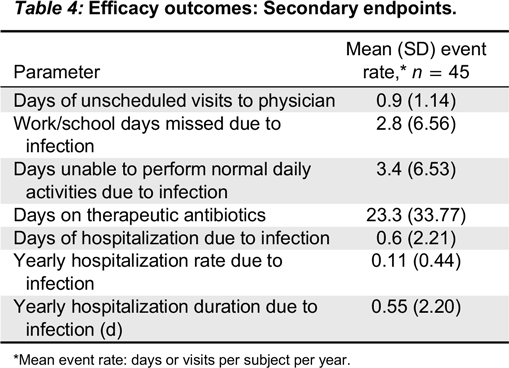
Kedrion 10% IVIG was found to be efficacious by meeting the primary study endpoint, which was less than 1.0 acute serious bacterial infection per subject per year. Two acute serious bacterial infections occurred during the entire study period and thus the upper limit of 99% confidence interval was 0.11. Secondary efficacy endpoints were also remarkable for low rates of missed work/school days. The mean rate for the subject missing work/school was 2.8 days per subject per year, which is comparable to reports describing other IVIG products (
Berger and Pinciaro 2004;
Berger 2007;
Stein et al. 2009;
Wasserman et al. 2012) and is lower than the average number missed days of work per year by US workers (
Centers for Disease Control and Prevention 2013;
Official Disability Guidelines 2013). The frequency of unscheduled visits to a physician’s office or emergency room were extremely low and far below the mean of 3.0 predicted in the normal population and below most published reports in IVIG trials. Very limited use of therapeutic antibiotics and a low rate of hospitalization were also recorded in this study.
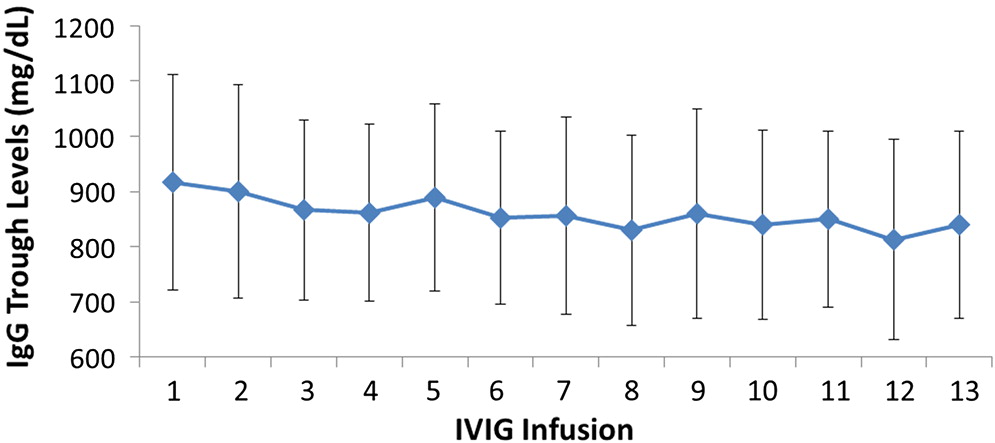
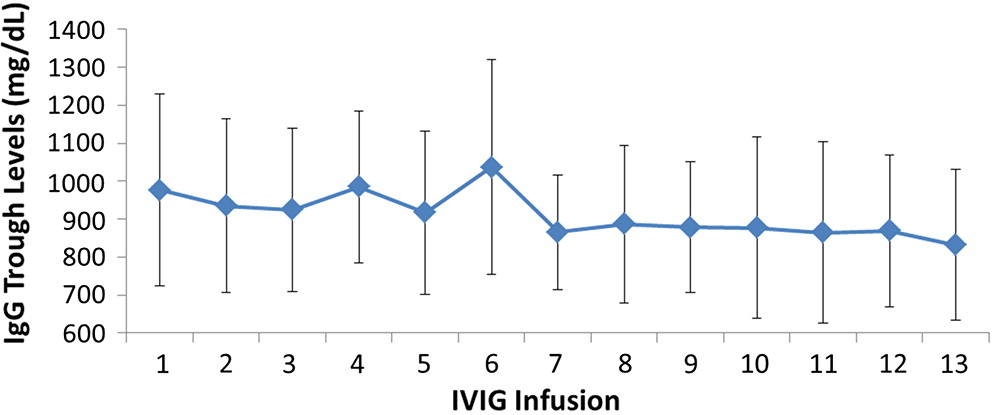
All patients had serum IgG trough levels higher than 500 mg/dL at the end of the study with mean concentrations of 843.7 ± 169.8 and a median of 828.0 mg/dL, a level shown to be extremely effective in preventing infections in patients with PID (
Roifman et al. 1987,
2003). PK studies of total IgG, IgG subclasses, and specific antibodies confirmed the good bioavailability of Kedrion 10% IVIG.
In summary, this clinical trial enrolled subjects with confirmed PID and antibody deficiency. The results of excellent tolerability and efficacy of Kedrion IVIG 10% are therefore significant and compare favorably to other IVIG products currently available.