Heterozygous mutations in RelB can be associated with immune dysregulation and lymphoma
Abstract
Background: The nuclear factor kappa-B (NFκB) family of transcription factors is essential for numerous processes, including the development and function of the innate and adaptive immune response, inflammation and cell growth, differentiation, and survival. Recently, patients with homozygous mutations in the gene for the NFκB transcription factor RelB have been described as presenting with features of combined immunodeficiency such as recurrent infection and failure to thrive as well as reduced response to mitogens and an inability to maintain an adequate antibody response to immunizations.
Methods: The immune status and genetics of the parents of patients with homozygous RelB mutations were assessed. In vitro mitogen stimulation, flow cytometry, and cytokine ELISA were used to assess immunological status and signal transduction pathways.
Results: Four patients were confirmed to have heterozygous RelB mutations. The majority of patients had evidence of immune dysfunction with impaired in vitro responses to PHA and antigens. One patient developed lymphoma.
Conclusion: Heterozygous RelB mutations can be associated with immune dysregulation with impaired mitogen and antigen responses and lymphoma. It is likely that the immune defects apparent in RelB deficient humans are due to a wider effect of RelB on the classical NFκB pathway (involving RelA and c-Rel) through cross-regulation of activation and expression in addition to RelB’s function within the alternate pathway.
Statement of novelty: We describe for the first time the immune abnormalities in patients with heterozygous RelB mutations.
Introduction
The nuclear factor kappa-B (NFκB) family of transcription factors is essential for numerous processes, including the development and function of the innate and adaptive immune response, inflammation and cell growth, differentiation, and survival (Sun 2011). The NFκB signaling pathway consists of 5 major proteins including NFκB 1 p50/p105, NFκB 2 p52/p100, RelA/p65, RelB, and c-Rel (Sun 2011; Millet et al. 2013). These proteins function as dimeric transcription factors that regulate the expression of genes influencing a range of processes including innate and adaptive immunity. The NFκB pathway is divided into 2 main signaling pathways, the classical or canonical pathway mediated by RelA and the alternative or noncanonical pathway mediated by RelB (Sun 2011). The canonical signaling pathway is initiated by a surface receptor, such as TLR4, TNFR, and TCR, causing activation of the IKK complex (IKKβ, IKKα, NEMO), which in turn phosphorylates IκB proteins leading to their proteasomal degradation and release of RelA and p50 heterodimers (Millet et al. 2013). The RelA and p50 heterodimers then translocate into the nucleus where they bind NFkB sites and induce target gene expression (Millet et al. 2013).
The noncanonical pathway is activated through extracellular signals, such as LTβR, CD40L, BAFF, and TWEAK, which cause TRAF3 degradation in the proteosome and stabilization of NIK (Millet et al. 2013). IKKα is then activated, resulting in the proteosomal processing of p100, converting it to p52 (Millet et al. 2013). This creates transcriptionally competent NFκB p52/RelB complexes that translocate to the nucleus and induce target gene expression (Millet et al. 2013). In addition to its receptor activated functions, the presence of RelB is proposed to be necessary for the regulation of other family members (Oeckinghaus et al. 2011; Sun 2011; Millet et al. 2013). RelB knockout mice show that the protein plays an important role in the development of lymphoid tissue as knockout mice often lack splenic and thymic structures, including the germinal centers, marginal zones, and thymic medulla (Burkly et al. 1995; Weih et al. 1995). Furthermore, RelB knockout mice often die early of severe multisystem inflammatory syndrome with T-cell and monocytic infiltrates in multiple organs (Burkly et al. 1995).
Primary immunodeficiency can result from mutations at various stages in the NFκB pathway. Specifically, mutations in NFĸB Essential Modulator (NEMO, also known as inhibitor factor of nuclear factor kappa-B kinase subunit gamma (IKKγ)), IĸBα, and inhibitor of NFĸ-B kinase subunit beta (IKKβ, also known as IKK2) have been described (Doffinger et al. 2001; Courtois et al. 2003; Janssen et al. 2004; Orange et al. 2004; Lopez-Granados et al. 2008; Picard et al. 2011; Pannicke et al. 2013; Schimke et al. 2013). Recently, Merico et al. (2015) described homozygous mutations in the gene encoding RelB (c.1191 C > A; Y397 stop) resulting in the premature stop and the ablation of RelB expression. The 3 patients described had features of combined immunodeficiency with repeated infections and failure to thrive. Immune assessment revealed normal to increased numbers of circulating lymphocytes that were unresponsive to mitogens or antigens in vitro and poor specific antibody response. Further studies reported that these patients highlighted that absence of RelB expression lead to intrinsic defects in both T and B lymphocyte maturation and function resulting in combined immunodeficiency and autoimmunity (Sharfe et al. 2015).
Here we present the parents of the 3 patients previously described with homozygous RelB mutations and highlight that heterozygous mutations in RelB can be associated with immune dysregulation and lymphoma.
Methods
Subjects
Subjects include the parents of patients recently described with homozygous RelB mutations (Merico et al. 2015). Patient data were compiled prospectively and retrospectively from medical records and were entered into the Canadian Centre for Primary Immunodeficiency Registry and tissue bank, which was approved by the SickKids Research Ethics Board (protocol # 1000005598). This included consent for patients and parents for genetic analysis, immune evaluation, collection of tissue and permission to publish.
T-cell proliferative responses
Lymphocyte proliferative responses to mitogens (including phytohemagglutinin (PHA) and anti-CD3 antibodies) and to a panel of recall antigens (including candida, tetanus, herpes zoster, and cytomegalovirus) were determined by thymidine incorporation at day 3 or day 6. All assays were performed in triplicate and were compared with simultaneously stimulated random normal controls.
Immunoglobulin and specific antibody determinations
Serum concentrations of immunoglobulins were measured by nephelometry. Serum IgE concentration was measured by radioimmunoassay with the IgE PRIST kit (Pharmacia Diagnostics, Quebec, Canada). Levels of serum antibodies to tetanus were measured by ELISA.
Cytokine secretion
Peripheral blood mononuclear cells (PBMCs) were cultured with PHA for 24 and 48 hours and culture supernatants collected for analysis of IL2 and IFNγ by ELISA. Kits were from R&D Systems Inc. (Minneapolis, MN).
Results
Case reports
Father 1 (F1) and Mother 1 (M1) are both of Irish descent and are the parents of patients 1 and 2 as described in Merico et al. (2015). Medical history was remarkable for asthma in F1 and psoriasis and atopic dermatitis in M1.
Father 2 (F2) and Mother 2 (M2) are also of Irish descent and are the parents of patient 3 described in Merico et al. (2015) and Sharfe et al. (2015). M2 has an unremarkable past medical history. F2 had a diagnosis of asthma as a child. At the age of 27 years, F2 presented with soft tissue mass in the right supraclavicular area. CT scan of his neck, chest, abdomen, and pelvis reported multiple pathologically enlarged lymph nodes along the parotid chain. Excisional biopsy confirmed Hodgkin’s lymphoma, nodular sclerosing subtype. Bone marrow biopsy had normal trilineage hematopoietic marrow with no morphologic evidence of Hodgkin’s lymphoma. He was managed with chemotherapy and has been in remission for 2 years. Family history was remarkable for a first cousin who also had Hodgkin’s lymphoma successfully treated with chemotherapy and a grandmother and grandfather with colon cancer.
Evaluation of the immune system
All patients displayed normal numbers of circulating white blood cells (Table 1). Lymphocyte immunophenotyping revealed that circulating lymphocyte numbers were normal to elevated in all 4 patients (Table 1). In vitro responses to PHA and antigens were low in all patients except M2. F2 and M2 had appropriate levels of immunoglobulins and response to vaccines (vaccine response was not measured in F1 and M1).
Table 1:
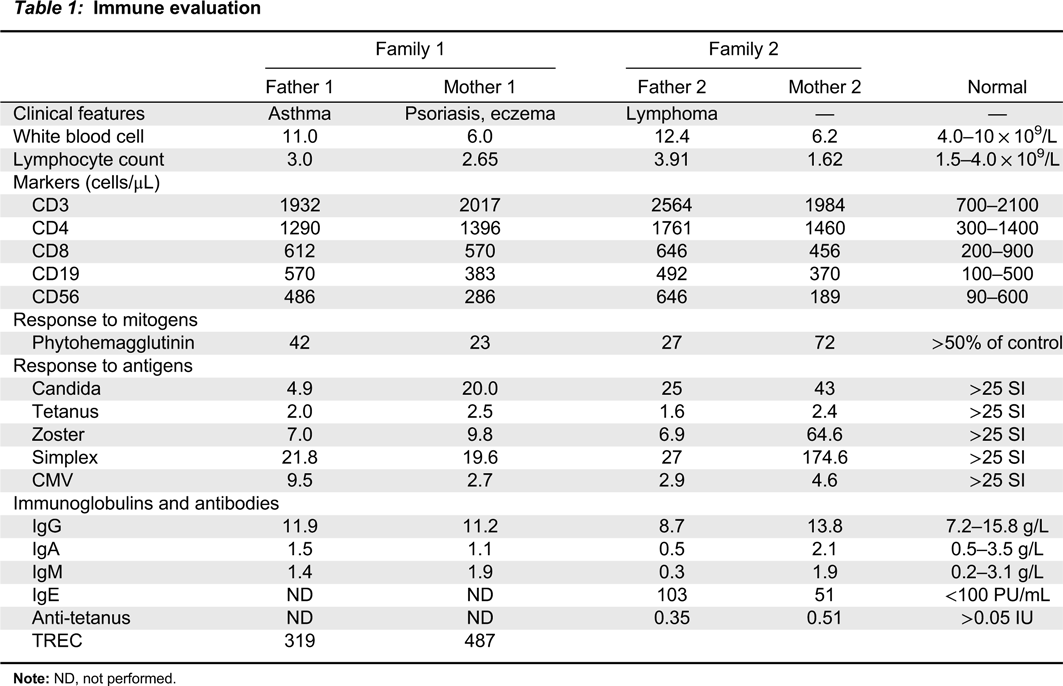
Note: ND, not performed.
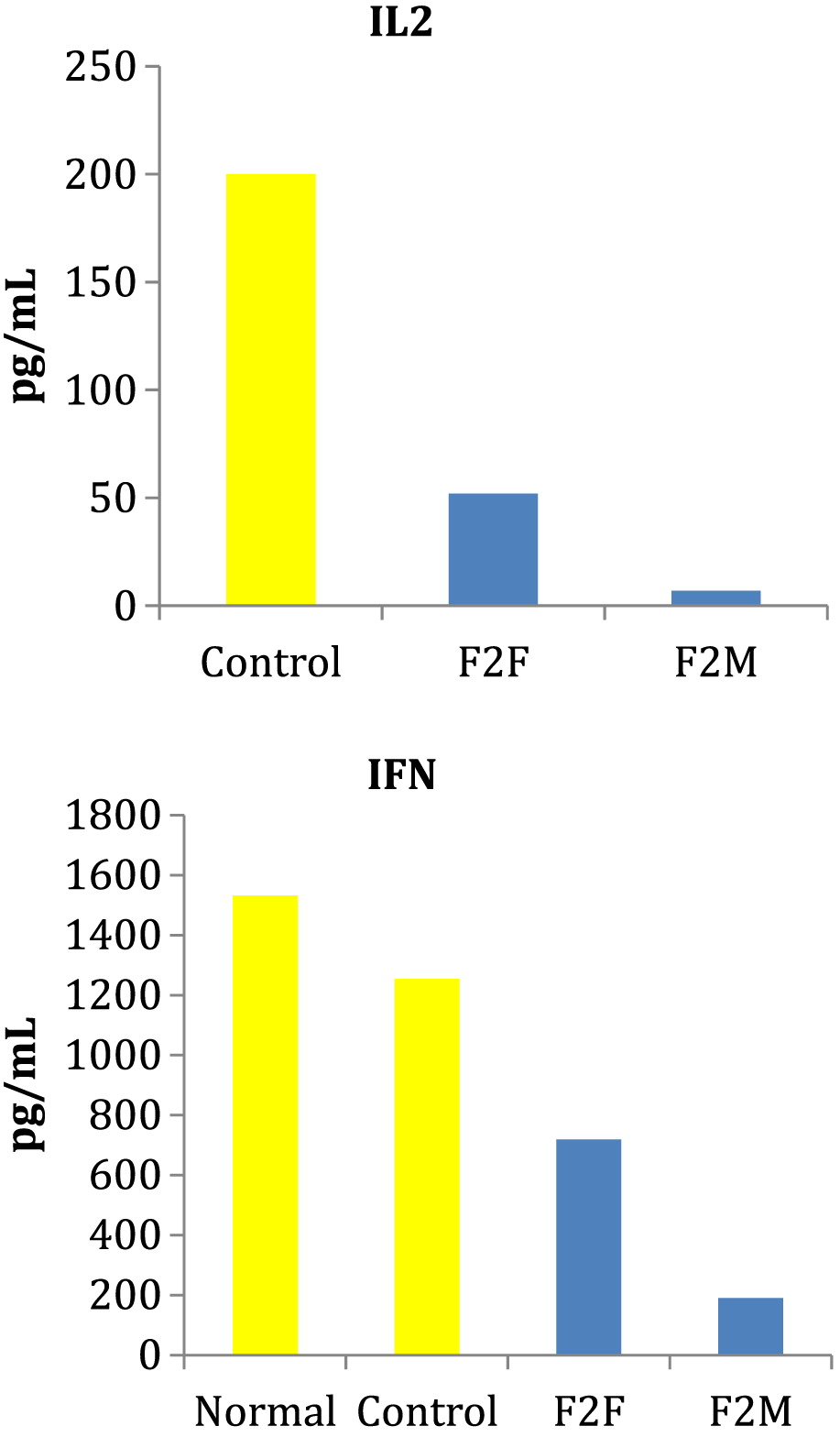
Cytokine secretion
PBMCs were cultures with PHA for 24 and 48 hours and culture supernatants collected for analysis of IL2 and IFNγ by ELISA. Patients heterozygous for the RelB mutation had markedly reduced PHA induced production of both IL2 and IFNγ (Figure 1).
Figure 1:
Discussion
A novel type of combined immunodeficiency associated with a homozygous mutation in the RelB gene has recently been described (Merico et al. 2015; Sharfe et al. 2015). Here we describe the clinical and immunologic features of 4 patients, the parents of the homozygous RelB patients, all whom were discovered to have heterozygous RelB mutations. The 3 patients described with homozygous RelB mutations were identified as having a premature stop codon mutation leading to markedly decreased levels of RelB mRNA, presumably due to message instability (Merico et al. 2015). Furthermore, RelB protein was undetectable in these patient’s lymphocytes, suggesting that any potential fragment that may be translated is also unstable and subsequently degraded (Merico et al. 2015). Sharfe et al. (2015) detailed the effects of RelB deficiency on lymphocyte development and function (Sharfe et al. 2015). In particular, patients with homozygous RelB mutations have T-cell dysfunction as evident by markedly dysplastic thymus with reduced production of T cells, abnormal TCR repertoire with clonal expansion, depressed in vitro responses to T-cell mitogens and antigens, reduced production of IL2 and IFNγ, and exaggerated in vitro responses to anti-CD3 and CD28 (Sharfe et al. 2015). Furthermore, B-cell dysfunction was demonstrated with reduced BAFF receptor expression, absent memory/mature B cells and low to absent T-cell dependent antibody responses (Sharfe et al. 2015). The result of the intrinsic defects in both T- and B-lymphocyte maturation and function was a presentation consistent with combined immunodeficiency with features including repeated infections and failure to thrive. The 4 patients described here with heterozygous RelB mutations did not have recurrent infections as a clinical feature but did have evidence of immune dysregulation. Specifically, our patients also had impaired T-cell function with inadequate response to mitogens and antigens as well as reduced IL2 and IFNγ production following stimulation with PHA. These findings suggest that heterozygous RelB mutations also result in deficient T lymphocytes in vivo. The lack of recurrent infections in our patients may reflect residual RelB function and (or) the involvement of compensatory mechanisms protective against infection in the group with heterozygous RelB mutations.
Impaired immune functioning is also evident by the lymphoma that developed in F2. Aberrant NFκB activity has been observed in many cancers, including both solid and hematopoietic malignancies (Basseres and Baldwin 2006; Naugler and Karin 2008). Evidence suggests that NFκB pathways may mediate pro-apoptotic effect, growth arrest, and inhibition of cancer development (Perkins 2004; Chen and Castranova 2007). Specifically, a recent study demonstrated that RelB provides cell proliferation-inhibitory signals in murine fibroblasts and that RelB ectopic expression inhibits xenograft tumor growth in vivo, whereas RelB knockdown enhances it; Jacque et al. (2013) proposed these findings were secondary to direct transcriptional activation of the p53 tumor-suppressor gene by RelB. These findings highlight the importance of malignancy surveillance in this patient population.
Conclusion
Here we highlighted that heterozygous RelB mutations can be associated with immune dysregulation and lymphoma. It is likely that the immune defects apparent in RelB-deficient humans are due to a wider effect of RelB on the classical NFκB pathway (involving RelA and c-Rel) through cross-regulation of activation and expression, in addition to RelB’s function within the alternate pathway (Sharfe et al. 2015).
Acknowledgements
This work was supported by the Program for Immunogenomics and the Canadian Centre for Primary Immunodeficiency, the Jeffrey Modell Foundation, and Immunodeficiency Canada. The Centre for Applied Genomics at SickKids was supported by Genome Canada through the Ontario Genomics Institute, Canada Foundation for Innovation, and the Ontario Ministry of Research and Innovation.
REFERENCES
Basseres D.S. and Baldwin A.S.2006. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene.25:6817–6830.
Burkly L., Hession C., Ogata L., Reilly C., Marconi L.A., Olson D., Tizard R., Cate R., and Lo D.1995. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature.373(6514):531–536.
Courtois G., Smahi A., Reichenbach J., Doffinger R., Cancrini C., Bonnet M., Puel A., Chable-Bessia C., Yamaoka S., Feinberg J., Dupuis-Girod S., Bodemer C., Livadiotti S., Novelli F., Rossi P., Fischer A., Israel A., Munnich A., Le Deist F., and Casanova J.L.2003. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Invest.112(7), 1108–1115.
Chen F. and Castranova V.2007. Nuclear factor-kappaB, an unappreciated tumor suppressor. Cancer Res.67:11093–11098.
Doffinger R., Smahi A., Bessia C., Geissmann F., Feinberg J., Durandy A., Bodemer C., Kenwrick S., Dupuis-Girod S., Blanche S., Wood P., Rabia S.H., Headon D.J., Overbeek P.A., Le Deist F., Holland S.M., Belani K., Kumararatne D.S., Fischer A., Shapiro R., Conley M.E., Reimund E., Kalhoff H., Abinun M., Munnich A., Israel A., Courtois G., and Casanova J. L.2001. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet.27(3):277–285.
Jacque E., Billot K., Authier H., Bordereaux D., and Baud V.2013. RelB inhibits cell proliferation and tumor growth through p53 transcriptional activation. Oncogene.32:2661–2669.
Janssen R., van Wengen A., Hoeve M.A., ten Dam M., van der Burg M., van Dongen J., van de Vosse E., van Tol M., Bredius R., Ottenhoff T.H., Weemaes C., van Dissel J.T., and Lankester A.2004. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes. J. Exp. Med.200(5):559–568.
Lopez-Granados E., Keenan J.E., Kinney M.C., Leo H., Jain N., Ma C.A., Quinones R., Gelfand E.W., and Jain A.2008. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat.29(6):861–868.
Merico D., Sharfe N., Hu P., Herbrick J., and Roifman C.M.2015. RelB deficiency causes combined immunodeficiency. LymphoSign J.2(3):147–155.
Millet P., McCall C., and Yoza B.2013. RelB: An outlier in leukocyte biology. J. Leukoc. Biol.94(5):941–951.
Naugler W.E. and Karin M.2008. NF-kappaB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev.18:19–26.
Oeckinghaus A., Hayden M.S., and Ghosh S.2011. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol.12(8):695–708.
Orange J.S., Jain A., Ballas Z.K., Schneider L.C., Geha R.S., and Bonilla F.A.2004. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J. Allergy. Clin. Immunol.113(4):725–733.
Pannicke U., Baumann B., Fuchs S., Henneke P., Rensing-Ehl A., Rizzi M., Janda A., Hese K., Schlesier M., Holzmann K., Borte S., Laux C., Rump E.M., Rosenberg A., Zelinski T., Schrezenmeier H., Wirth T., Ehl S., Schroeder M.L., and Schwarz K.2013. Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med.369(26):2504–2514.
Perkins N.D.2004. NF-kappaB: tumor promoter or suppressor?Trends Cell Biol.14:64–69.
Picard C., Casanova J.L., and Puel A.2011. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin. Microbiol. Rev.24(3):490–497.
Schimke L.F., Rieber N., Rylaarsdam S., Cabral-Marques O., Hubbard N., Puel A., Kallmann L., Sombke S.A., Notheis G., Schwarz H.P., Kammer B., Hokfelt T., Repp R., Picard C., Casanova J.L., Belohradsky B.H., Albert M.H., Ochs H.D., Renner E.D., and Torgerson T.R.2013. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy. J. Clin. Immunol.33(6):1088–1099.
Sharfe N., Merico D., Karanxha A, Macdonald C., Dadi H., Ngan B., Herbrick J., and Roifman C.M.2015. The effects of RelB deficiency on lymphocyte development and function. J. Autoimmun.65:90–100.
Sun S.C.2011. Non-canonical NF-kappaB signaling pathway. Cell. Res.21(1):71–85.
Weih F., Carrasco D., Durham S.K., Barton D.S., Rizzo C.A., Ryseck R.P., Lira S.A., and Bravo R.1995. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell.80(2):331–340.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 2 • June 2016
Pages: 55 - 60
History
Received: 17 November 2015
Accepted: 17 December 2015
Accepted manuscript online: 13 January 2016
Version of record online: 13 January 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AlisonHaynes, PalinderKamra, and ChaimRoifman. 2016. Heterozygous mutations in RelB can be associated with immune dysregulation and lymphoma. LymphoSign Journal.
3(2): 55-60. https://doi.org/10.14785/lymphosign-2015-0014
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
