Review of hereditary angioedema
Abstract
Hereditary angioedema (HAE) is a rare disease caused by deficiency of C1 esterase inhibitor (C1-INH). It is an autosomal dominant disease caused by a variety of mutations in the C1-INH gene. C1-INH is an important regulator of several pathways. One pathway it affects is the kallikrein–kinin pathway, which results in the generation of bradykinin. Bradykinin is an important mediator of edema. Diagnosis is based on low levels of C1-INH. HAE with normal C1-INH is also recognized in the literature and the pathophysiology is due to another aspect of the pathway being affected leading to increased bradykinin level. Bradykinin results in intermittent swelling of the cutaneous and mucosal surfaces. The swelling usually evolves over several hours and lasts a few days. Location of the swelling can involve any part of the body including fatal laryngeal edema. Newer treatments exist to treat acute attacks and reduce the frequency of future attacks. Earlier diagnosis and treatment of hereditary angioedema can prevent HAE-associated mortality.
Statement of novelty: New treatments are used to treat these attacks. These treatments are aimed at patients having a more normal life with hereditary angioedema.
Introduction
Hereditary angioedema (HAE) was first described by William Osler in 1888. However, it was not until 1963 that Donaldson and Evan first described the biochemical abnormality responsible for HAE, the absence of C1 esterase inhibitor (C1-INH) in patients with the disease (Cicardi et al. 2014).
Traditionally, the literature has described two types of HAE, Type I and Type II. Both are autosomal-dominant disorders resulting from mutation in the C1-INH gene on chromosome 11. More than 200 mutations in the gene have been described (Kaplan 2010). There has been no correlation found between genotype and clinical phenotype in Type I and Type II HAE (Caccia et al. 2014).
Type I HAE accounts for approximately 80%–85% of all cases. The mutations in Type I HAE can occur anywhere in the gene. Abnormal proteins that are either misfolded or truncated are inefficiently secreted. This results in decreased antigenic levels and functional activity of the C1-INH protein (Zuraw 2008).
Type II HAE mutations, about 15%–20% of all HAE cases, usually involve single amino acid substitutions, mainly at exon 8 at or near the active site. Levels of circulating C1-INH are usually normal. The secreted protein is dysfunctional, resulting in normal antigenic C1-INH levels but low function C1-INH (Zuraw 2008).
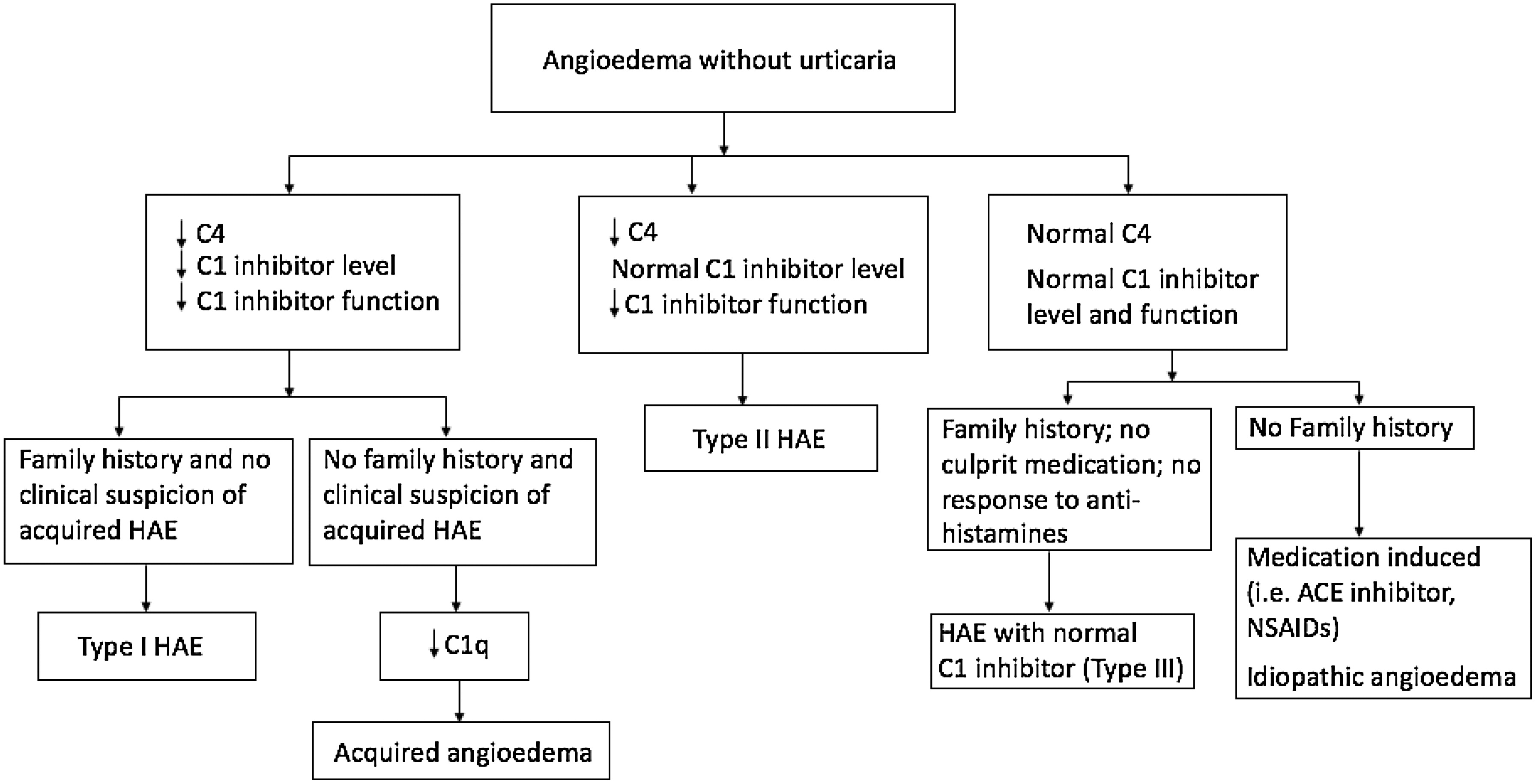
In addition to Type I and II, another type of hereditary angioedema is recognized. It has been called HAE with normal C1-INH, Type III HAE, or estrogen-dependent hereditary angioedema. Hereditary angioedema with normal C1-INH manifests with sporadic recurrent angioedema but normal C1-INH level and function. The diagnosis of HAE with normal C1-INH is made with meeting the following criteria: (i) a history of recurrent angioedema in the absence of concomitant hives or concomitant use of a medication known to cause angioedema; (ii) documented normal or near normal C4, C1-INH level, and C1-INH function; and (iii) demonstration of a Factor XII mutation that is associated with the disease or a positive family history of angioedema and documented evidence of lack of efficacy of chronic high dose antihistamine therapy (cetirizine at 40 mg/day or the equivalent, for at least 1 month and an interval expected to be associated with 3 or more attacks of angioedema) (Zuraw et al. 2012). In some cases, bradykinin accumulation appears dependent on Factor XII. The majority of patients do not have any well-described mutations; a small percentage of patients have mutations in Factor XII. This type of angioedema appears to predominantly affect females in high estrogen states. The postulated mechanism by which estrogen exacerbates angioedema is by increasing multiple components of the kallikrein–kinin system (Duan et al. 2009; Riedl 2013; Levy et al. 2014).
Acquired angioedema can also be secondary to a malignancy (usually lymphoproliferative disorders) and autoimmune diseases and present with identical clinical and lab profiles as HAE.
Although not discussed in this review of hereditary angioedema, it is important to ask about medications including angiotensin converting enzyme inhibitor and nonsteroidal anti-inflammatory drug use in patients presenting clinically with angioedema without urticaria.
The prevalence of Type I and II HAE is uncertain but is estimated to be approximately 1 case per 50,000 persons, without known differences among ethnic groups (Zuraw 2008).
Pathophysiology
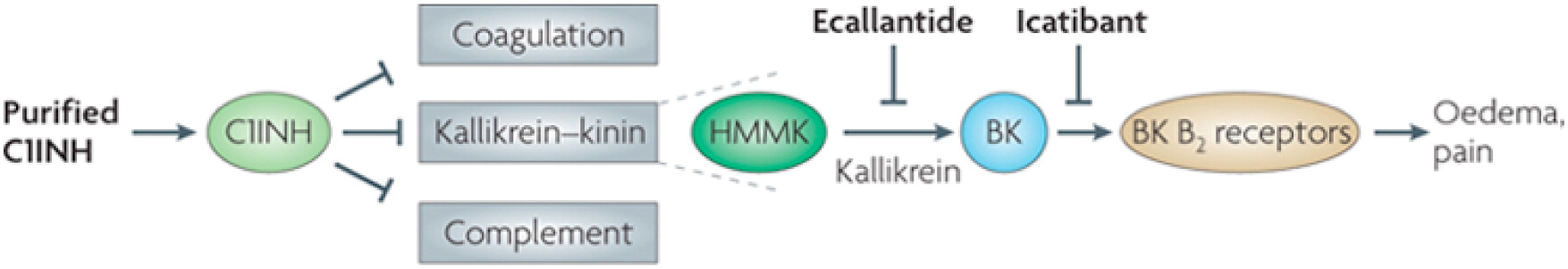
In Type I HAE and Type II HAE, there is a deficiency of C1-INH, a protein serine protease inhibitor that is involved in the complement, coagulation, and contact pathways (Longhurst and Cicardi 2012). It has been shown that an overproduction of bradykinin, a nonapeptide, in the contact pathway is the main mediator of the increased vascular permeability in Type I and Type II HAE (Cugno et al. 2009). The deficiency in C1-INH decreases the normal inhibition of kallikrein (Longhurst and Cicardi 2012), which enables greater cleavage of high molecular-mass kininogen by kallikrein and a resulting increased generation of bradykinin (Cugno et al. 2009). Abnormally high plasma levels of bradykinin can increase swelling by binding to its cognate receptors (the bradykinin B2 receptor) on vascular endothelial cells, thereby increasing tissue permeability, vascular dilation, and vascular smooth muscle relaxation (Cugno et al. 2009). Figure 1 shows a simplified diagram of C1-INH.
Figure 1:
Clinical manifestations
Age of onset
Type I and Type II HAE usually presents in childhood, sometimes as early as 2 years of age (Zuraw 2008). Usual symptom onset occurs from 8 to 12 years (Farkas et al. 2007), with an increase of attacks often during puberty (Kaplan 2010). Bork et al. (2015) found that in HAE patients with normal C1-INH, those with mutations in the Factor XII gene, have an average age of onset at 20.3 years, whereas those without Factor XII mutations have an average onset of symptoms at 29.6 years.
Duration
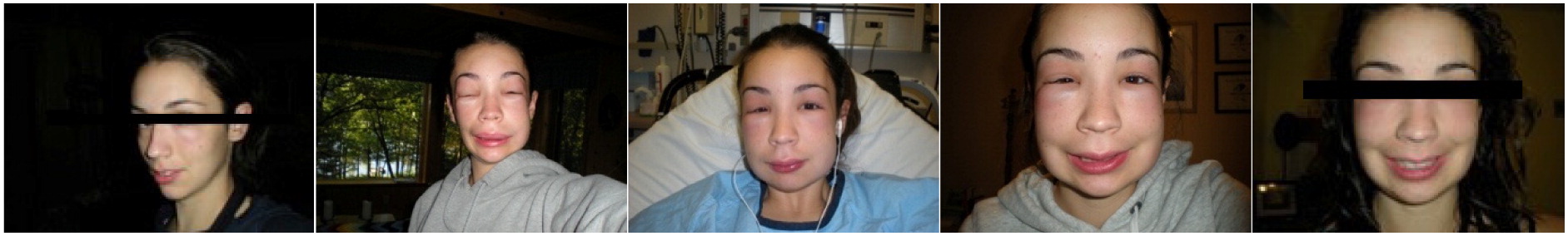
The swelling classically worsens slowly but relentlessly over the first 24 hours, then gradually subsides over the subsequent 48–72 hours (Zuraw 2008). A sequential progression of an attack is shown in Figure 2.
Figure 2:
Frequency of attacks
Untreated patients have attacks every 7–14 days on average, with the frequency ranging from every 3 days to virtually never (Zuraw 2008).
Features of attacks
HAE is typically characterized by recurrent episodes of nonpruritic, nonpitting, subcutaneous, or submucosal edema of the arms, legs, hands, feet, trunk, bowels, genitalia, face, tongue, or larynx (Zuraw 2008). Swelling in the absence of urticaria is suggestive of HAE. In about 40%–87% of HAE patients there are prodromal symptoms (up to 16 hours prior) including erythema marginatum, substantial fatigue, or local discomfort (Longhurst and Cicardi 2012). HAE patients with normal C1-INH do not have erythema marginatum but frequently have hemorrhaging into skin swellings (Bork 2010).
The site of swelling varies among patients and within patients between attacks (Kaplan 2010). Attacks may originate in one area and may spread to another (Zuraw 2008). HAE patients with normal C1-INH more frequently have skin swelling, tongue swelling, and abdominal pain attacks (Bork 2010).
Intra-abdominal swelling causes abdominal pain, distension, nausea, vomiting, and diarrhea. Symptoms are often mistaken for other causes of acute abdomen, and HAE patients often undergo unnecessary invasive investigations and surgeries (Longhurst and Cicardi 2012).
Laryngeal attacks pose the greatest risk for patients as they may progress to death from asphyxiation (Zuraw 2008).
Diagnosis
Plasma C4 levels are a valuable screening test for Type I and Type II HAE, with most of those affected having a reduced level between attacks (Zuraw et al. 1986) and nearly 100% having a low level during attacks (Lang et al. 2012). If C4 is low, further tests can distinguish Type I HAE patients who have low antigenic C1-INH levels and low functional C1-INH levels from Type II patients with normal antigenic C1-INH levels but low functional C1-INH levels. C1q and C3 are normal with Type I and Type II HAE patients (Zuraw 2008). Reduction in C4 and C1-INH levels and function of C1-INH should be confirmed one to three months after initial testing (Cicardi et al. 2014). Figure 3 shows a diagnostic algorithm.
Figure 3:
Two methods (chromogenic or immunoenzymatic) are available to measure C1-INH function. They are based on the measurement of the capacity of plasma to inhibit the esterase activity of a fixed amount of C1s. The chromogenic assay is usually preferred owing to a higher positive predictive value of close to 100%. It is important to send the C1-INH function to specialized laboratories to avoid inaccurate results. The samples should be stored at –20°C (Wagenaar-Bos et al. 2008). Genetic testing is not necessary for the diagnosis given the heterogeneity of mutations responsible for Type I and Type II HAE (Cicardi et al. 2014). Genetic testing is also not widely available. The average time between the onset of symptoms and diagnosis was 22 years as of 1977 and was still more than 10 years as of 2005 (Zuraw 2008).
The importance of diagnosis was shown in a study by Bork et al. (2012). Mortality by asphyxiation due to laryngeal angioedema was significantly higher in patients with undiagnosed HAE (63 cases) than in patients diagnosed with HAE (7 cases). HAE patients must be informed of the risks of laryngeal angioedema and be educated about emergency treatments and the need for early treatment administration (Bork et al. 2012).
Treatment of Type I and Type II HAE
Acute attacks
Acute attacks may be spontaneous or triggered from external stimuli; they can be life threatening and involve airway compromise. The treatments listed in Table 1 have been used successfully to treat acute attacks. Plasma-derived C1-INH and icatibant are licensed in Canada for treatment of acute attacks. Plasma-derived C1-INH has been shown to decrease the severity and length of attacks. Icatibant is a synthetic 10-amino acid peptide that acts as a selective bradykinin B2 receptor antagonist. It has been shown to effectively treat HAE attacks (Betschel et al. 2014).
Table 1:
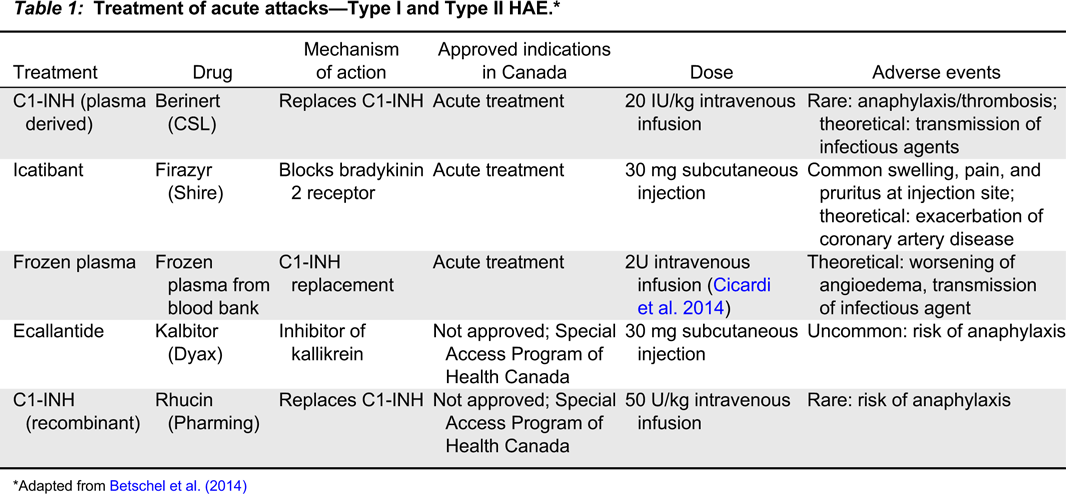
*
Adapted from Betschel et al. (2014)
Frozen plasma should be used only if other therapies are not available. Evidence for effective treatment is low and it contains potential substrates for the generation of additional bradykinin that, in theory, could worsen attacks of angioedema (Betschel et al. 2014).
Ecallantide and recombinant C1-INH have both been shown to be effective treatments for acute attacks. They are not approved by Health Canada but can applied for through the Special Access Program of Health Canada (Betschel et al. 2014).
Attenuated androgens and tranexamic acid should not be used to treat acute attacks, as there is a lack of evidence toward their effectiveness (Betschel et al. 2014).
Short-term prophylaxis
Prophylaxis for procedures and known patient-specific triggers is generally recommended. Dosages for plasma-derived C1-INH have not been established for prophylaxis. In Canada, Berinert is licensed for acute attacks and Cinryze is licensed for long-term prophylaxis. In Europe, Cinryze (1000 units) is licensed to be given within 24 hours of the procedure and Berinert (1000 units) is licensed for within 6 hours of an anticipated procedure (Betschel et al. 2014).
Danazol can be considered 5 days before the anticipated procedure or trigger and continued for 2–3 days after (Danazol 2.5–10 mg/kg/day, maximum 600 mg/day).
Anti-fibrinolytic agents (i.e., tranexamic acid) have also been used at 25 mg/kg 2–3 times daily to a maximum of 3–6 g/day, 5 days before and 2–5 days after a procedure or anticipated trigger. This agent should only be used if other therapies are not available, as its efficacy for prevention of attacks is not well studied (Betschel et al. 2014).
Long-term prophylaxis
Patients with recurrent episodes of angioedema may be considered for long-term prophylaxis to reduce the frequency, duration, and severity of attacks (Table 2).
Table 2:
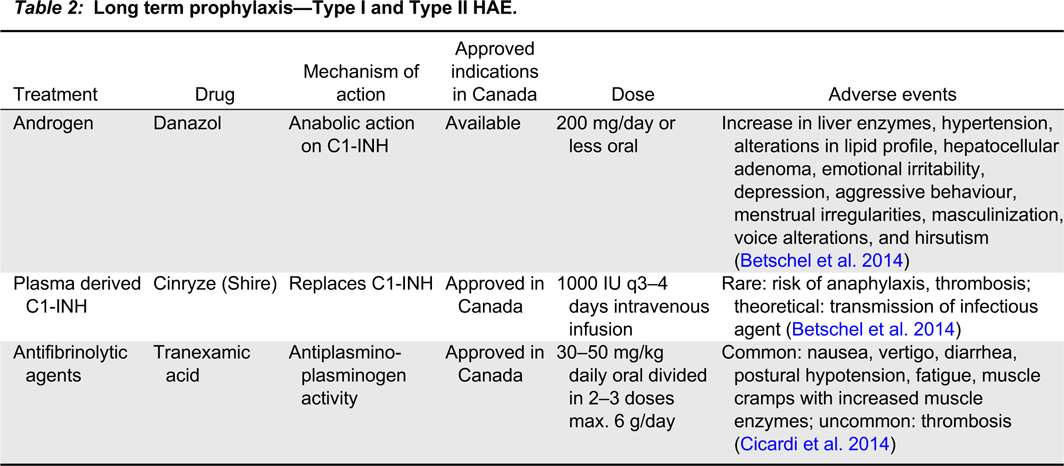
Long-term prophylaxis aims to reduce the frequency and severity of attacks to improve quality of life. Controlled trials and observational studies have demonstrated that prophylaxis with 17-alpha-alkylated anabolic androgens reduced the frequency and severity of attacks, although there are many side effects and a need for monitoring. Plasma-derived C1-INH (Cinryze) is approved for use in Canada. It has been shown to be effective in reducing the number, duration, and severity of attacks (Betschel et al. 2014). Anti-fibrinolytic agents have also been used in long-term prophylaxis of HAE attacks; they have been used when androgens were contraindicated (Kaplan 2010) and they can be used in children for whom treatment with androgens could affect growth (Longhurst and Cicardi 2012). Tranexamic acid for long-term prophylaxis can be given at doses of 20–40 mg/kg/day in 2 or 3 divided doses (maximum dose: 3 g/day in 2 or 3 divided doses per day) (Farkas et al. 2007).
Treatment of HAE with normal C1-INH
There is not enough evidence to support a specific treatment of acute attacks or for prophylaxis in patients with HAE with normal C1-INH. Some experts feel that a trial of effective treatments demonstrated for Type I and Type II may be appropriate (Betschel et al. 2014).
Conclusion
Hereditary angioedema is a life-threatening disease that is under-recognized and under-diagnosed. There have been many advances in specific treatments in the last few years for HAE. Given the mortality of undiagnosed disease, it is very important for the medical community to recognize and appropriately diagnose HAE. Effective treatment should be made available to HAE patients for acute attacks and long term prophylaxis when appropriate.
REFERENCES
Betschel S., Badiou J., Binkley K., Hébert J., Kanani A., Keith P., Lacuesta G., Yang B., Aygören-Pürsün E., Bernstein J., Bork K., Caballero T., Cicardi M., Craig T., Farkas H., Longhurst H., Zuraw B., Boysen H., Borici-Mazi R., Bowen T., Dallas K., Dean J., Lang-Robertson K., Laramée B., Leith E., Mace S., McCusker C., Moote B., Poon M.C., Ritchie B., Stark D., Sussman G., and Waserman S.2014. Canadian hereditary angioedema guideline. Allergy Asthma Clin. Immunol.10(1):50.
Bork K.2010. Diagnosis and treatment of hereditary angioedema with normal C1inhibitor. Allergy Asthma Clin. Immunol.6(1):15.
Bork K., Hardt J., and Witzke G.2012. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J. Allergy Clin. Immunol.130(3):692–697.
Bork K., Wulff K., Witzke G., and Hardt J.2015. Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy.70(8):1004–1012.
Caccia S., Suffritti C., and Cicardi M.2014. Pathophysiology of hereditary angioedema. Pediatr. Allergy Immunol. Pulmonol.27(4):159–163.
Cicardi M., Aberer W., Banerji A., Bas M., Bernstein J.A., Bork K., Caballero T., Farkas H., Grumach A., Kaplan A.P., Riedl M.A., Triggiani M., Zanichelli A., and Zuraw B.2014. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the Hereditary Angioedema International Working Group. Allergy.69(5):602–616.
Cugno M., Zanichelli A., Foieni F., and Cicardi M.2009. C1-inhibitor deficiency and angioedema: Molecular mechanisms and clinical progress. Trends Mol. Med.15(2):69–78.
Duan Q.L., Binkley K., and Rouleau G.A.2009. Genetic analysis of Factor XII and bradykinin catabolic enzymes in a family with estrogen-dependent inherited angioedema. J. Allergy Clin. Immunol.123(4):906–910.
Farkas H., Varga L., Szeplaki G., Visy B., Harmat G., and Bowen T.2007. Management of hereditary angioedema in pediatric patients. Pediatrics.120(3):e713–e722.
Kaplan A.P.2010. Enzymatic pathways in the pathogenesis of hereditary angioedema: The role of C1 inhibitor therapy. J. Allergy Clin. Immunol.126(5):918–919.
Lang D., Aberer W., Bernstein J., Chng H.H., Sevciovic Grumach A., Hide M., Maurer M., Weber R., and Zuraw B.2012. International consensus on hereditary and acquired angioedema. Ann. Allergy Asthma Immunol.109(6):395–402.
Levy J., Rivard G.E., Wagner E., Beezhold D., Berlin N., Fan L., Zhang Z., and Sussman G.L.2014. Examination of genetic variants involved in generation and biodisposition of kinins in patients with angioedema. Allergy Asthma Clin. Immunol.10(1):60.
Longhurst H. and Cicardi M.2012. Hereditary angio-oedema. Lancet.379(9814):474–481.
Riedl M.A.2013. Hereditary angioedema with normal C1-INH (HAE type III). J. Allergy Clin. Immunol. Pract.1(5):427–432.
Wagenaar-Bos I.G., Drouet C., Aygören-Pursun E., Bork K., Bucher C., Bygum A., Farkas H., Fust G., Gregorek H., Hack C.E., Hickey A., Joller-Jemelka H.I., Kapusta M., Kreuz W., Longhurst H., Lopez-Trascasa M., Madalinski K., Naskalski J., Nieuwenhuys E., Ponard D., Truedsson L., Varga L., Nielsen E.W., Wagner E., Zingale L., Cicardi M., and van Ham S.M.2008. Functional C1-inhibitor diagnostics in hereditary angioedema: Assay evaluation and recommendations. J. Immunol. Methods.338(1–2):14–20.
Zuraw B.L.2008. Hereditary angioedema. N. Engl. J. Med.359(10):1027–1036.
Zuraw B.L., Bork K., Binkley K.E., Banerji A., Christiansen S.C., Castaldo A., Kaplan A., Riedl M., Kirkpatrick C., Magerl M., Drouet C., and Cicardi M.2012. Hereditary angioedema with normal C1 inhibitor function: Consensus of an international expert panel. Allergy Asthma Proc.33(Suppl 1):S145–S156.
Zuraw B.L., Sugimoto S., and Curd J.G.1986. The value of rocket immunoelectrophoresis for C4 activation in the evaluation of patients with angioedema or C1-inhibitor deficiency. J. Allergy Clin. Immunol.78(6):1115–1120.
Zuraw B.L., Yasothan U., and Kirkpatrick P.2010. Ecallantide. Nat. Rev. Drug. Discov.9(3):189–190.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 2 • June 2016
Pages: 47 - 53
History
Received: 17 January 2016
Accepted: 3 May 2016
Accepted manuscript online: 10 May 2016
Version of record online: 10 May 2016
Copyright
© 2016 © Agricultural Institute Of Canada.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Lisa W.Fu, TamlynFreedman-Kalchman, StephenBetschel, and GordonSussman. 2016. Review of hereditary angioedema. LymphoSign Journal.
3(2): 47-53. https://doi.org/10.14785/lymphosign-2016-0001
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
