IPEX syndrome caused by a novel mutation in FOXP3 gene can be cured by bone marrow transplantation from an unrelated donor after myeloablative conditioning
Abstract
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome is a rare primary immunodeficiency caused by inherited defects in the FOXP3 gene that impair regulatory T cells. IPEX syndrome can be cured by hematopoietic stem cell transplantation (HSCT) from HLA-matched unrelated donors (MUD); however, the best conditioning prior to HSCT for IPEX syndrome is not known.
Here we report on a patient suffering from IPEX syndrome, including immune-mediated colitis and membranous nephropathy, without polyendocrinopathy, caused by a novel mutation in the Forkhead domain of the FOXP3 gene. The patient's symptoms resolved following MUD HSCT after myeloablative conditioning performed at 16 months of age. The patient is clinically well, 3 years after HSCT, with robust immune reconstitution and fully engrafted. The lack of extensive autoimmune damage might have contributed to the patient's favourable outcome following MUD HSCT with myeloablative conditioning.
Statement of novelty: We describe a novel mutation in the FOXP3 gene causing IPEX syndrome and the correction of IPEX syndrome with bone marrow transplant from a HLA-matched unrelated donor following myeloablative conditioning.
Introduction
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, first described in 1982, is a rare and life-threatening primary immunodeficiency characterized by widespread autoimmunity (Powell et al. 1982). Most patients present in the first months of life with severe colitis, failure to thrive, insulin-dependent diabetes mellitus, and eczematous dermatitis. Other manifestations include lymphadenopathy, splenomegaly, and various autoimmune abnormalities including hemolytic anemia, autoimmune thrombocytopenia, hypothyroidism, hepatitis, arthritis, and alopecia (Wildin et al. 2002; Van der Vliet and Nieuwenhuis 2007).
IPEX syndrome results from defects in regulatory T cells (Treg), which account for 5%–10% of peripheral CD4+ T cells (Piccirillo et al. 2008). Treg are characterized by cell surface expression of CD4 and CD25 as well as intracellular expression of FOXP3. Mutations in the FOXP3 gene have been identified as the cause for IPEX syndrome (Bennet et al. 2001a, 2001b). The FOXP3 gene, located at position Xp11.23–Xq13.3, encodes a protein called Scurfin, a transcriptional regulator that is crucial for the generation of Treg. FOXP3 has a proline rich N-terminal domain that is important for regulating FOXP3 expression, a C2H2 zinc finger motif, and a leucine zipper domain implicated in the formation of homo- and heterodimers (Verbsky and Chatila 2013). Additionally, FOXP3 contains a winged-helix domain, also known as the “Forkhead box” or FKH domain, critical for the DNA binding and suppression properties of FOXP3. In a recent review, more than 50 unique mutations, spanning the entire coding and noncoding regions of the FOXP3 gene were reported, primarily involving the FKH domain (D'Hennezel et al. 2012). Some genotype–phenotype correlation has been suggested in IPEX syndrome, including the presence and function of Treg (Zennaro et al. 2011).
IPEX syndrome is often fatal by 2 years of age, mainly because of intractable diarrhea, complications of diabetes, hemorrhage, or sepsis (Wildin et al. 2002). Management involves immune suppression with steroids, calcineurin, and mTOR inhibitors as well as monoclonal antibodies against T cells; however, these treatments often provide only partial and temporary benefit (Di Rocco and Marta 1996; Bindl et al. 2005; McGinness et al. 2006; Taddio et al. 2007; Yong et al. 2008). Since 2000, increasing numbers of patients with IPEX syndrome have received hematopoietic stem cells transplantations (HSCT) from HLA-matched related or matched unrelated donors (MUD; Baud et al. 2001; Wildin et al. 2002; Mazzolari et al. 2005; Lucas et al. 2006; Rao et al. 2007; Gambineri et al. 2008; Zhan et al. 2008; Dorsey et al. 2009; Seidel et al. 2009; Burroughs et al. 2010; Moes et al. 2010; Kasow et al. 2011). In contrast to patients with severe combined immune deficiency who lack T cells where HSCT without any myeloablation can be effective, patients with IPEX syndrome might require elimination of their T cells prior to HSCT to ensure long-term engraftment. However, as many of patients who received MUD HSCT using full myeloablative conditioning died from various complications, there is debate about this approach for patients suffering from IPEX syndrome (Baud et al. 2001; Wildin et al. 2002)
Here we report on a patient with IPEX syndrome caused by a novel FOXP3 mutation in the FKH domain who was cured by a myeloablative MUD bone marrow transplant.
Case report
The male patient was born at term to nonconsanguineous parents of English descent after a normal pregnancy and delivery. From 2 months of age, the patient suffered intractable profuse watery and bloody diarrhea with up to 30 bowel movements a day. There was no improvement in stooling despite changes in formula feeds, performed because of presumed food allergy. At 2 months of age the patient experienced respiratory syncytial virus bronchiolitis that resolved. At 8 months of age the patient experienced rotavirus enteritis. The patient was admitted to hospital at 9 months of age for protracted diarrhea. He was found to suffer from failure to thrive, hypertension, extensive eczema, lymphadenopathy, ascites, hypotonia, and motor developmental delay. There was no history of immunodeficiency, autoimmunity, or bowel disease in the parents or their extended families, apart from a maternal second cousin with type 1 diabetes mellitus.
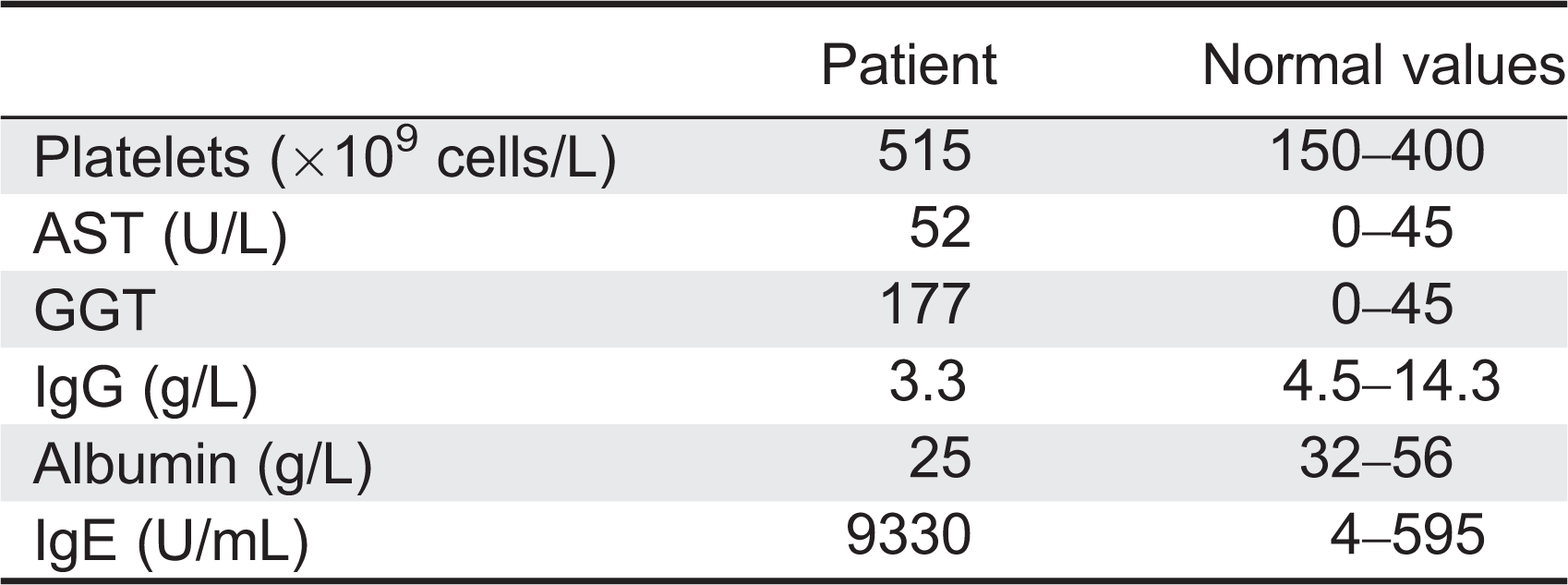
Laboratory investigations at admission revealed hypoalbuminemia, massive proteinuria, and microscopic hematuria. Complete blood count was normal apart from elevated platelet count (Table 1). Liver enzymes were mildly deranged. Flow cytometry analysis showed normal numbers of CD20+, CD3+, CD4+, CD8+, and NK cells. T-cell receptor excision circles, indicative of new thymus emigrants, were within normal range for the age. In-vitro responses of lymphocytes to stimulations with mitogens and antigens, serum IgA and IgM levels, as well as specific antibody titers were within normal ranges. Serum IgG was slightly reduced, possibly reflecting the protein loss, whereas IgE concentration was markedly elevated (Table 1). Neutrophil oxidative burst index and complement levels were also normal. Auto-antibodies against islet cells, glutamic acid decarboxylase (GAD), insulin, thyroid peroxidase (TPO), thyroid receptor, adrenal, nuclear, smooth muscle, mitocondrial, and liver–kidney microsome (LKM) were all negative. Duodenal biopsies showed complete villous atrophy, crypt hyperplasia, and an inflammatory infiltrate in the lamina propria (Figure 1A) as well as loss of the normal villus brush border and absence of goblet cells (Figure 1B). Crypt abscesses with apoptotic epithelial cells were evident (Figure 2A), whereas the lamina propria infiltrate consisted predominantly of lymphocytes with occasional plasma cells and eosinophils (Figure 2B). Immuno-staining for CD3 demonstrated an extensive T-cell infiltrate (Figure 3A) with the presence of FOXP3+ cells (Figure 3B). These findings suggested an autoimmune colitis. Biopsies from stomach, colon, sigmoid, and rectum were unremarkable. A renal biopsy showed membranous glomerulonephritis (Figure 4). Sanger sequencing of the FOXP3 gene revealed a novel hemizygous T1117A substitution (Figure 5) putatively causing an exchange of isoleucine for phenylalanine at amino acid 373, an area in the FKH domain prone to mutations (D'Hennezel et al. 2012). The patient's mother was heterozygous for the mutation.
Figure 1:
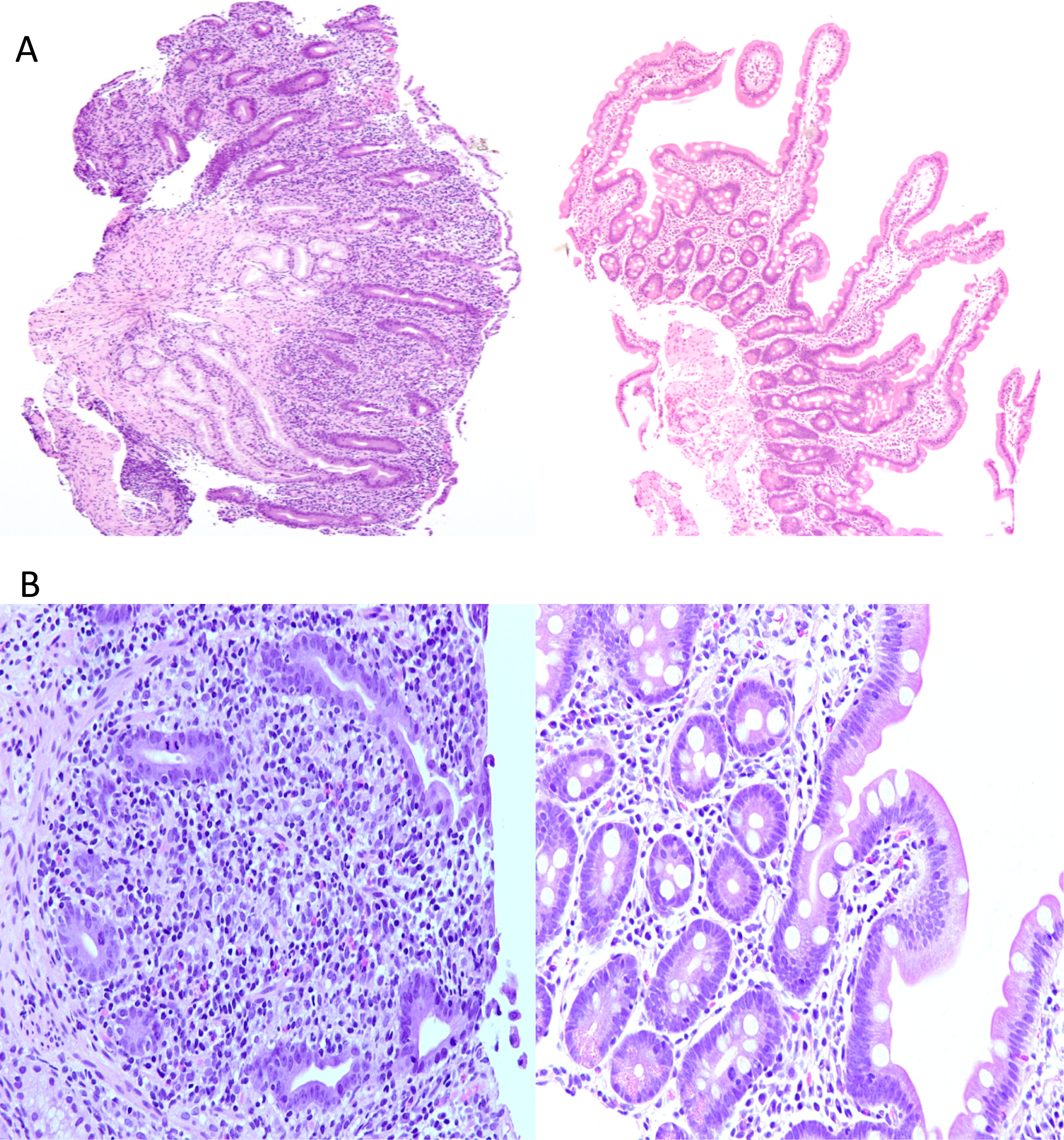
Figure 2:
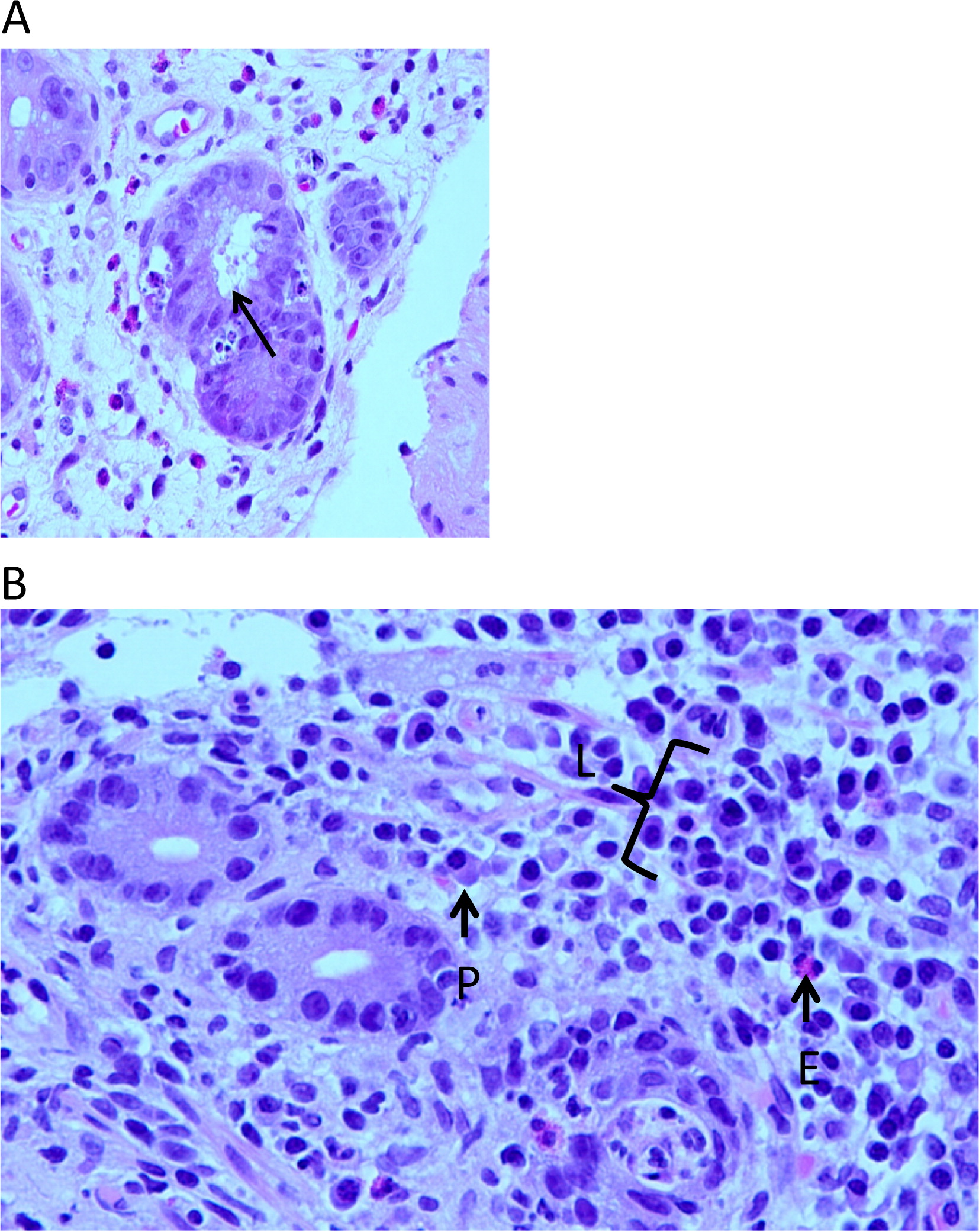
Figure 3:
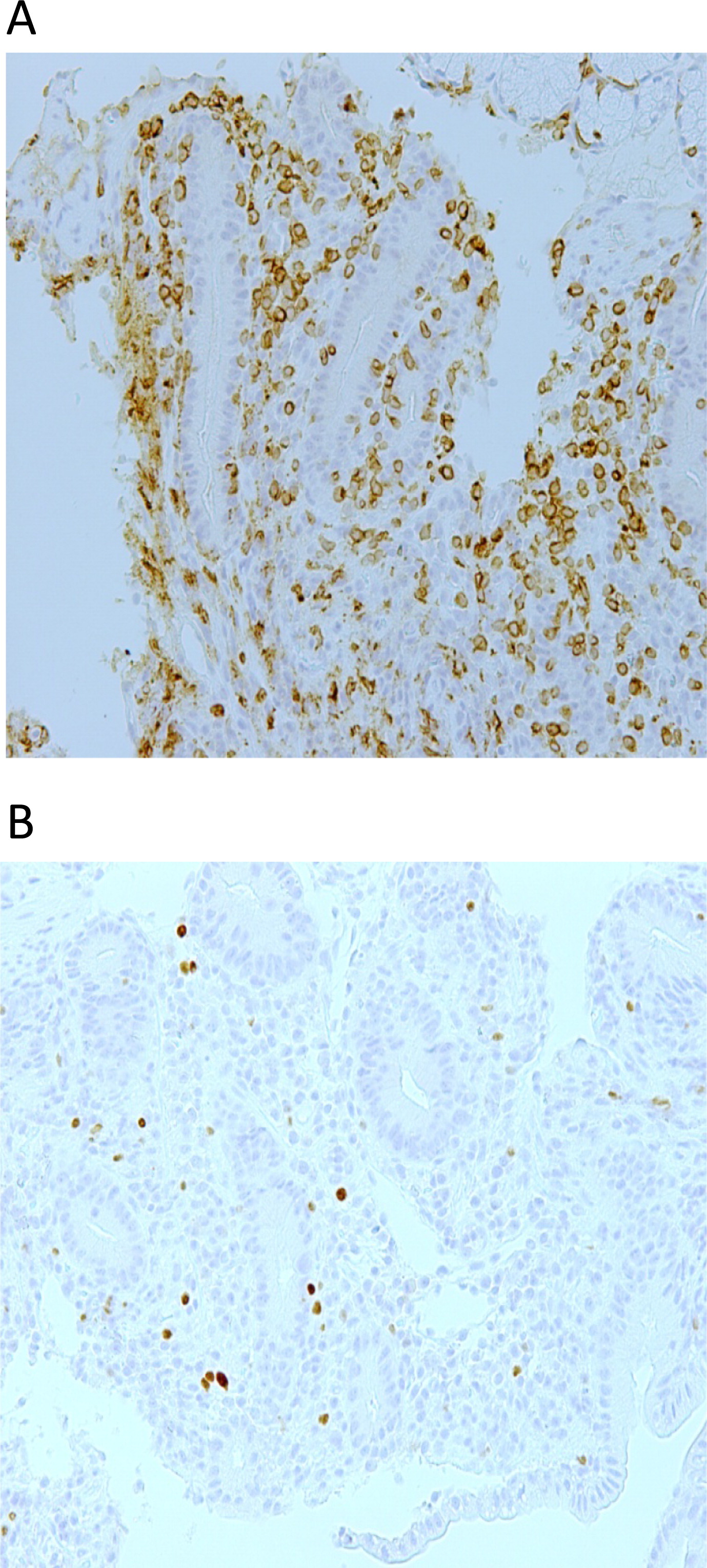
Figure 4:
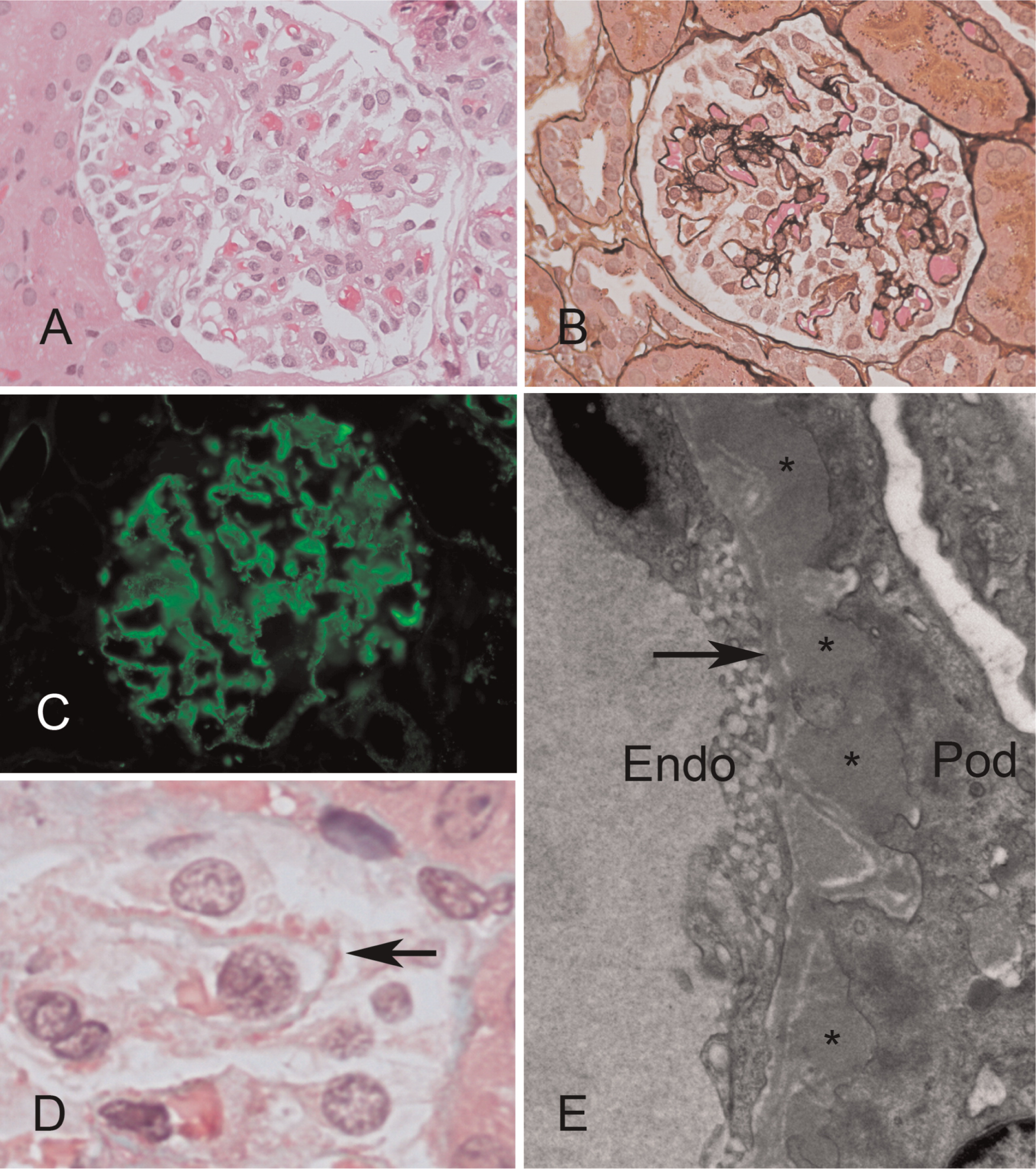
Figure 5:
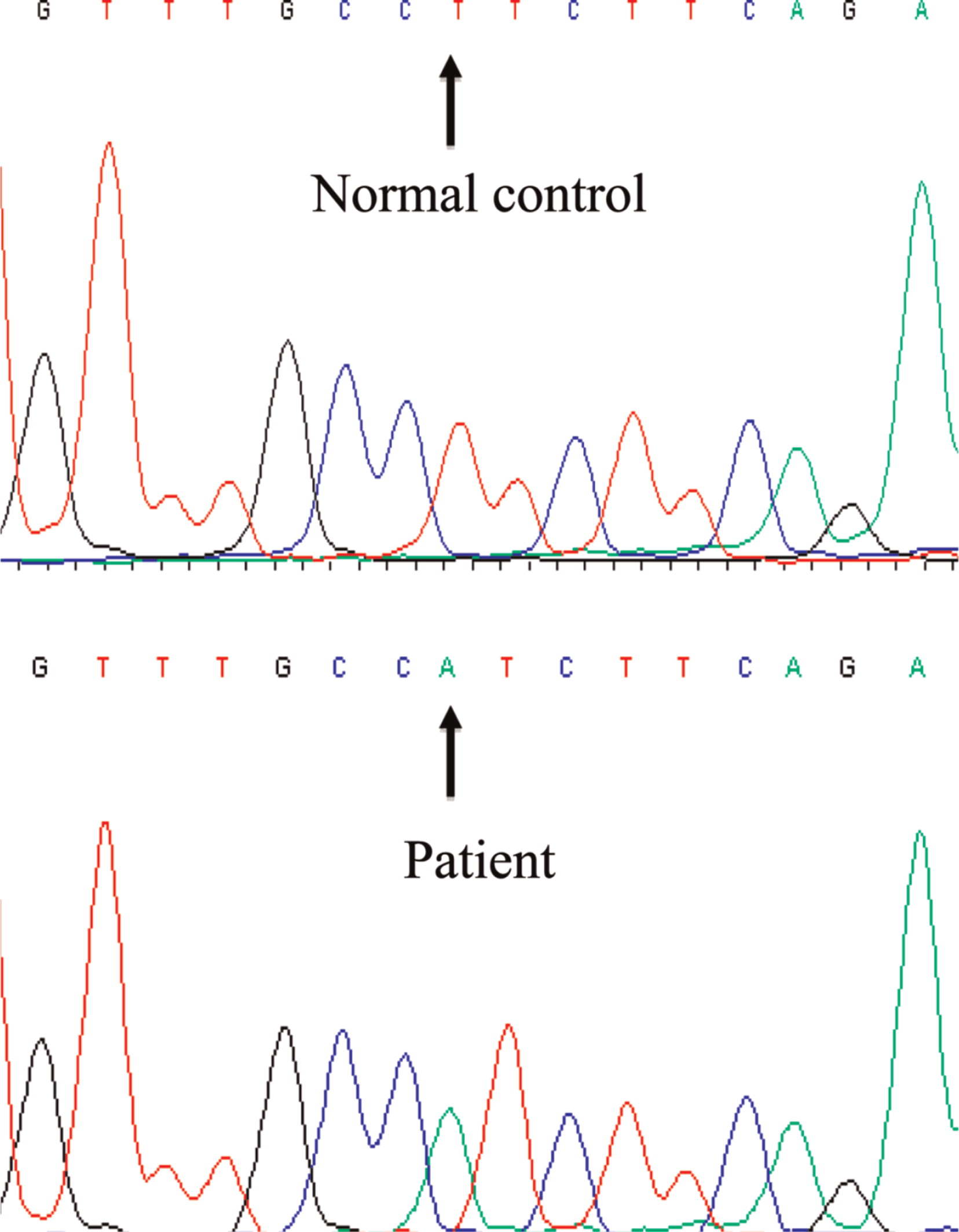
Table 1:
Despite prolonged immune suppression with methylprednisolone and cyclosporine, the diarrhea and proteinuria persisted. At 16 months of age the patient received bone marrow HSCT from a female who was CMV negative and 10 out of 10 HLA MUD. Conditioning consisted of 5 mg/kg busulfan for 4 days followed by 50 mg/kg cyclophosphamide for 4 days, a regimen similar to that used by us for other forms of primary immune deficiencies (Grunebaum et al. 2006). The patient tolerated the conditioning regimen well except for transient elevation of liver transaminases. The patient developed grade IV skin graft-versus-host disease despite methylprednisolone and cyclosporine prophylaxis that was treated with methylprednisolone pulse followed by mycophenolate mofetil and sirolimus. The diarrhea improved with reduced frequency of stools within 1 month of HSCT and normalization of stool frequency and consistency a month later. The nephropathy improved dramatically; the urinary albumin–creatinine ratio declined from a high of 1139.5 at presentation (normal < 3.5) to 12 within 3 months of HSCT and was normalized by 9 months.
At 3.5 years after HSCT, the patient is eating normal solid foods without any evidence of colitis. There are also no indications of nephritis, endocrinopathy, autoimmunity, neurological abnormalities, or developmental delay. He has not had any significant infections, lymphocyte sub-populations numbers are normal, and FISH analysis with X and Y chromosome probes of the patient's lymphocytes demonstrates complete (100%) donor chimerism.
Discussion
Patients with IPEX syndrome typically present in infancy with severe colitis and endocrinopathy (Wildin et al. 2002). The patient presented here suffered predominantly from severe diarrhea; however, he did not have any endocrine involvement, which might have delayed his diagnosis. Indeed, several experienced pediatricians suspected initially that the patient had a food allergy, emphasizing the need for increased alertness for the possibility of immune dysregulation among infants with nonresolving colitis (Montalto et al. 2009; Patey-Mariaud de Serre et al. 2009). In addition, our patient exhibited membranous glomerulonephritis, an uncommon nephropathy in childhood that has been infrequently recognized among the various kidney abnormalities associated with FOXP3 deficiency (Moudgil et al. 2007; Hashimura et al. 2009; Rodrigo et al. 2013).
The number of FOXP3-positive T cells in the patient's gut biopsy appeared normal, indicating that the novel T1117A mutation did not prevent protein translation and expression. Indeed, in many other patients with IPEX syndrome, mutations in the FKH domain, which likely impair nuclear import and DNA binding capacity, did not abrogate FOXP3 expression (D'Hennezel et al. 2012). Consequently, as demonstrated in our patient, immunodetection of FOXP3 in biopsies should not exclude IPEX syndrome.
It is customary to use substantial conditioning before HSCT of patients with immunodeficiency that still possess significant residual immune function. This is done to allow for rapid and sustained long-term allograft survival and recapitulation of cellular and humoral immunity. In FOXP3 deficiency, although deregulated, most T- and B-cell functions are preserved, thus conditioning prior to stem cell infusion is required. The first 3 patients who received HSCT for FOXP3 deficiency were prepared with complete myeloablative conditioning (Baud et al. 2001; Wildin et al. 2002). Unfortunately none survived the procedure, consequently deterring further use of myeloablative conditioning in IPEX syndrome. The 3 patients died from hemophagocytic lymphohistiocytosis, pneumonitis, and post-transplant lymphoproliferative disorder at 25 months, 6 months, and 3 months, respectively, after HSCT. However, none of these deaths seemed to be directly related to toxicity of busulfan or cyclophosphamide. Moreover, at least 1 other patient who received full myeloablative conditioning survived (Mazzolari et al. 2005), suggesting that other factors such as extensive comorbidities including autoimmunity might have affected the outcome. Accordingly, we estimated that the risk of graft failure and the need for repeated HSCT associated with reduced intensity conditioning outweighs the risk of full myleoablative conditioning. Indeed we found no evidence for busulfan or cyclophosphamide toxicity in our patient. Moreover, the patient's colitis and nephritis resolved rapidly while immune reconstitution and lymphoid engraftment was robust. Thus, our data suggest that the quest for modified nonmyeloablative conditioning should be carefully studied using factual, rather than perceived, myeloablation toxicity in HSCT for immunodeficiency.
In conclusion, vigilance for the possibility of IPEX syndrome in infants with colitis should remain high, even in the presence of FOXP3+ cells. Also it will be beneficial in the future to compare long term survival, engraftment, and immune reconstitution among patients with FOXP3 deficiency who underwent HSCT following myeloablative versus reduced intensity conditioning.
Acknowledgements
The authors thank Dr. Roifman for performing the molecular analysis of the FOXP3 gene. EG is supported by the Donald and Audrey Chair for Immunology Research, Hospital for Sick Children and the University of Toronto, Toronto, Ontario.
REFERENCES
Baud O., Goulet O., Canioni D., Le Deist F., Radford I., Rieu D., Dupuis-Girod S., Cerf-Bensussan N., Cavazzana-Calvo M., Brousse N., Fischer A., and Casanova J.L. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marro transplantation N. Eng. J. Med. 2001 334 1758 -1762
Bennet C.L., Brunkow M.E., and Ramsdell F. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA->AAUGAA) leads to the IPEX syndrome Immunogenetics. 2001a 53 435 -439
Bennet C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., and Ochs H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3 Nat. Genet. 2001b 27 1 20 -21
Bindl L., Torgerson T., Perroni L., Youssef N., Ochs H.D., Goulet O., and Ruemmele F.M. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome) J. Pediatr. 2005 147 2 256 -259
Burroughs L.M., Torgerson T.R., Storb R., Carpenter P.A., Rawlings D.J., Sanders J., Scharenberg A.M., Skoda-Smith S., Englund J., Ochs H.D., and Woolfrey A.E. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome J. Allergy. Clin. Immunol. 2010 126 5 1000 -1005
D'Hennezel E., Dhuban K.B., Torgerson T., and Piccirilo C. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome J. Med. Genet. 2012 49 291 -302
Di Rocco M. and Marta R. X linked immune dysregulation, neonatal insulin dependent diabetes, and intractable diarrhea Arch. Dis. Child. Fetal. Neonatal. Ed. 1996 75 2 F144
Dorsey M.J., Petrovic A., Morrow M.R., Dishaw L.J., and Sleasman J.W. FOXP3 expression following bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning Immunol. Res. 2009 44 1-3 179 -184
Gambineri E., Perroni L., Passerini L., Bianchi L., Doglioni C., Meschi F., Bonfanti R., Sznajer Y., Tommasini A., Lawitschka A., Junker A., Dunstheimer D., Heidemann P.H., Cazzola G., Cipolli M., Friedrich W., Janic D., Azzi N., Richmond E., Vignola S., Barabino A., Chiumello G., Azzari C., Roncarolo M.-G., and Bacchetta R. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity J. Allergy. Clin. Immunol. 2008 122 6 1105 -1112.e1
Grunebaum E., Grunebaum E., Mazzolari E., Porta F., Dallera D., Atkinson A., Reid B., Notarangelo L.D., and Roifman C.M. Bone marrow transplantation for severe combined immune deficiency JAMA. 2006 Feb 1 295 5 508 -518
Hashimura Y., Nozu K., Kanegane H., Miyawaki T., Hayakawa A., Yoshikawa N., Nakanishi K., Takemoto M., Iijima K., and Matsuo M. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome Pediatr. Nephrol. 2009 24 6 1181 -1186
Kasow K.A., Morales-Tirado V.M., Wichlan D., Shurtleff S.A., Abraham A., Persons D.A., and Riberdy J.M. Therapeutic in vivo selection of thymic-derived natural T regulatory cells following non-myeloablative hematopoietic stem cell transplant for IPEX Clin. Immunol. 2011 141 2 169 -176
Lucas K.G., Ungar D., Comito M., Bayerl M., and Groh B. Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome Bone Marrow Transplant. 2006 39 1 55 -56
Mazzolari E., Forino C., Fontana M., D'Ippolito C., Lanfranchi A., Gambineri E., Ochs H., Badolato R., and Notarangelo L.D. A new case of IPEX receiving bone marrow transplantation Bone Marrow Transplant. 2005 35 10 1033 -1034
McGinness J.L., Bivens M.M., Greer K.E., Patterson J.W., and Saulsbury F.T. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is associated with pemphigoid nodularis: a case report and review of literature J. Am. Acad. Dermatol. 2006 55 143 -148
Moes N., Rieux-Laucat F., Begue B., Verdier J., Neven B., Patey N., Torgerson T.T., Picard C., Stolzenberg M., Ruemmele C., Rings E.H.H.M., Casanova E.H., Piloquet J., Biver H., Breton A., Ochs A., Hermine H.D., Fischer O., Goulet A., Cerf–Bensussan O., and Ruemmele F.M. Reduced expression of FOXP3 and regulatory T-cell function in severe forms of early-onset autoimmune enteropathy Gastroenterology. 2010 139 3 770 -778
Montalto M., D'Onofrio F., Santoro L., Gallo A., Gasbarrini A., and Gasbarrini G. Autoimmune enteropathy in children and adults Scand. J. Gastroenterol. 2009 44 9 1029 -1036
Moudgil A., Perriello P., Loechelt B., Przygodzki R., Fitzerald W., and Kamani N. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy Pediatr. Nephrol. 2007 22 10 1799 -1802
Patey-Mariaud de Serre N., Canioni D., Ganousse S., Rieux-Laucat F., Goulet O., Ruemmele F., and Brousse N. Digestive histopathological presentation of IPEX syndrome Modern Pathol. 2009 22 1 95 -102
Piccirillo C.A., d'Hennezel E., Sgouroudis E., and Yurchenko E. CD4+ Foxp3+ regulatory T cells in the control of autoimmunity: in vivo veritas Curr. Opin. Immunol. 2008 20 6 655 -662
Powell B.R., Buist N.R.M., and Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy J. Pediatr. 1982 100 5 731 -737
Rao A., Kamani N., Filipovich A., Lee S.M., Davies S.M., Dalal J., and Shenoy S. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning Blood. 2007 109 1 383 -385
Rodrigo R., Atapattu N., and de Silva K. IPEX syndrome with membrano-proliferative nephrotic syndrome Ceylon. Med. J. 2013 58 1 43 -44
Seidel M.G., Fritsch G., Lion T., Jurgens B., Heitger A., Bacchetta R., Lawitschka A., Peters C., Gadner H., and Matthes-Martin S. Selective engraftment of donor CD4+25highFOXP3-positive T cells in IPEX syndrome after nonmyeloablative hematopoietic stem cell transplantation Blood. 2009 113 22 5689 -5691
Taddio A., Faleschini E., Valencic E., Granzotto M., Tommasini A., Lepore L., Andolina M., Barbi E., and Ventura A. Medium-term survival without haematopoietic stem cell transplantation in a case of IPEX: insights into nutritional and immunosuppressive therapy Eur. J. Pediatr. 2007 166 11 1195 -1197
Van der Vliet H.J.J. and Nieuwenhuis E.E. IPEX as a result of mutations in FOXP3 Clin. Dev. Immunol. 2007 2007 5 1 -5
Verbsky J.W. and Chatila T.A. Immune dysregulation, polyendocrinopathy, enteropathy,X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases Curr. Opin. Pediatr. 2013 25 6 708 -714
Wildin R.S., Smyk-Pearson S., and Filipovich A.H. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome J. Med. Genet. 2002 39 8 537 -545
Yong P.L., Russo P., and Sullivan K.E. Use of sirolimus in IPEX and IPEX-like children J. Clin. Immunol. 2008 28 5 581 -587
Zennaro D., Scala E., Pomponi D., Caprini E., Arcelli D., Gambineri E., Russo G., and Mari A. Proteomics plus genomics approaches in primary immunodeficiency: the case of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome Clin. Exp. Immunol. 2011 167 1 120 -128
Zhan H., Sinclair J., Adams S., Cale C.M., Murch S., Perroni L., Davies G., Amrolia P., and Qasim W. Immune reconstitution and recovery of FOXP3 (Forkhead Box P3)-expressing T cells after transplantation for IPEX (Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-linked) Syndrome Pediatrics. 2008 121 4 e998 -e1002
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: 31 - 38
History
Received: 27 August 2014
Accepted: 23 September 2014
Accepted manuscript online: 28 October 2014
Version of record online: 29 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Luis Murguia-Favela, Vy Hong-Diep Kim, Julia Upton, Paul Thorner, Brenda Reid, Adelle Atkinson, and Eyal Grunebaum. 2015. IPEX syndrome caused by a novel mutation in FOXP3 gene can be cured by bone marrow transplantation from an unrelated donor after myeloablative conditioning. LymphoSign Journal.
2(1): 31-38. https://doi.org/10.14785/lpsn-2014-0016
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
