Novel DOCK8 Mutation
Abstract
Background: Hyper IgE syndrome (HIES) is a primary immunodeficiency with sporadic, autosomal dominant (STAT3 mutation) and autosomal recessive (DOCK8 and TYK2 mutations) inheritance patterns. HIES secondary to DOCK8 mutation is characterized by extensive cutaneous viral and staphylococcal infections, recurrent sinopulmonary infections, severe allergic diseases, increased susceptibility to malignancy with lymphopenia, eosinophilia, and elevated immunoglobulin E (IgE). Methods: This case report highlights the clinical presentation and immune investigations of a male patient with a novel DOCK8 mutation. Results: Our patient presented with cutaneous viral infections including severe molluscum contagiosum and herpes simplex virus plus skin abscesses and acute otitis media. In addition to infections, he developed intermittent diarrhea, eczematous lesions, abnormal fingernails, oral ulcers, and Bell's palsy. Immune evaluation revealed lymphopenia, in particular low CD8 cells, low mitogen stimulation response, and poor specific antibody production requiring immunoglobulin replacement. Genetic sequencing confirmed a novel mutation in DOCK8. Conclusion: Patients with significant cutaneous viral and bacterial infections, recurrent sinopulmonary infections, severe allergic diseases, and lymphopenia with associated elevated IgE should be investigated for DOCK8 mutation. This case report highlights a novel mutation in the DOCK8 exon 45 aa1970, c.5908 G>C change alanine to proline homozygous change A1970 to P1970
Statement of novelty: This case reports on a novel mutation in DOCK8
Introduction
Hyper IgE syndrome (HIES) is classified as a well-defined syndrome with associated primary immunodeficiency (Al-Herz et al. 2011). HIES can be sporadic or familial, with both autosomal dominant and recessive cases described. The autosomal dominant form of HIES is most commonly caused by a mutation in signal transduction and activators of transcription 3 (STAT3) (Holland et al. 2007; Minegishi et al. 2007), whereas the less common autosomal recessive forms are linked to mutations in dedicator of cytokinesis 8 (DOCK8) (Engelhardt et al. 2009; Zhang et al. 2009) and tyrosine kinase 2 (TYK2) (Minegishi et al. 2006).
The human DOCK8 gene consists of 46–48 exons located on chromosome 9 (Su 2010) and is a member of the DOCK180 superfamily of guanine nucleotide exchange factors, which interacts with Rho GTPase (Ruusala and Aspenstrom 2004). The exact function of DOCK8 is unknown, but research supports its involvement in regulating cytoskeletal rearrangements that are essential for cellular structure, migration, and adhesion (Meller et al. 2005). Defects in DOCK8 have been shown to affect the immune system in numerous ways leading to a combined immunodeficiency. DOCK8 is involved in T-cell survival and the maintenance of CD8 T-cell memory (Lambe et al. 2011). Impaired T-cell activation (Zhang et al. 2009) and reduced numbers of T-cell excision circles (TRECS) suggest impaired thymopoiesis with possible restricted diversity of T-cell repertoire in the periphery (Dasouki et al. 2011). Furthermore, patients also have defective Th17 differentiation and long-term persistence (Al Khatib et al. 2009). Murine studies have shown that DOCK8 mutant B cells were unable to form marginal zone B cells or persist in germinal centers leading to impairment in affinity maturation of T-cell dependent antibody responses (Randall et al. 2009). It has also been described that Toll-like receptor 9 (TLR9) responses were considerably lower in DOCK8-deficient B cells and that DOCK8 functions as an adaptor in the TLR9–MyD88 signaling pathway in B cells (Jabara et al. 2012). Recently, DOCK8 deficiency has been shown to be associated with an increased production of autoantibodies, defective B cell tolerance, and quantitative and qualitative deficiencies in regulatory T cells (Janssen et al. 2014a). Lastly, the development and survival of mature natural killer T cells are impaired in DOCK8-deficient mice (Crawford et al. 2013).
HIES secondary to DOCK8 mutation has a more severe phenotype compared with the STAT3 mutations. Patients with DOCK8 mutations commonly present with significant viral cutaneous infections, sinopulmonary infections, and allergic diseases such as asthma, environmental allergies, and food allergies (Zhang et al. 2009; Alsum et al. 2013). This patient population lacks the extra immune abnormalities observed in STAT3 mutations, such as musculoskeletal and dental abnormalities and the characteristic coarse facial appearance. Early diagnosis of DOCK8 mutation patients is critical, as hematopoietic stem cell transplantation (HSCT) is offered to selected patients as a curative therapy.
Germline loss of function DOCK8 mutations found in patients with combined immunodeficiency were first described in 11 patients by Zhang et al. in 2009. Since this time other groups have been characterizing this primary immunodeficiency (Engelhardt et al. 2009; Al-Herz et al. 2012; Sanal et al. 2012; Alsum et al. 2013; Xue et al. 2014). We present here a novel case of DOCK8 mutation, including clinical presentation and immune investigation findings.
Results
A 13-year-old male patient from a nonconsanguineous Caucasian family was initially referred for intermittent diarrhea, recurrent molluscum contagiosum, skin abscesses, eczematous skin lesions, and lymphopenia. At 9 years of age, this patient developed diarrhea occurring every 3–4 months, lasting for 1 week, with as many as 10 episodes per day. The diarrhea episodes were associated with severe abdominal cramping and mucus with absence of blood in stools or associated fever. Infectious history was significant for severe molluscum contagiosum to the trunk and extremities beginning at childhood. Additional viral infections included extensive herpes simplex virus infection limited to the skin at 23 years of age. A history of bacterial infections was significant for 2 skin abscesses located on the left arm managed with incision and drainage at 13 years of age, and acute otitis media managed with oral antibiotics at 15 years of age. In addition to cutaneous infections, he had widespread nummular eczematous lesions over his extremities, trunk, and scalp beginning at 7 years of age. These lesions were initially diagnosed as psoriasis and managed with topical corticosteroids with minimal benefit. He had abnormal fingernails described as pitted and hypertrophied with associated peeling and crusting around the nailbed. Additional significant findings included intermittent oral ulcers and left-sided Bell's palsy. There was no history of allergic diseases.
Family history included a healthy nonconsanguineous mother and father, both from the Netherlands. He has an older sister who is healthy. The paternal grandmother had extensive atopic dermatitis. On the maternal side there was a female first cousin with vitilago and another first female cousin with leukemia diagnosed at 12 years of age.
Initial immune investigations were completed at 13 years of age (Tables 1 and 2). There was evidence of poor specific antibody response to tetanus (pre-vaccine titer 0.55 IU/mL, post-vaccine titer 0.85 IU/mL) and pneumococcus (post-vaccine titer 31.2 mg/L). Based on the poor specific antibody response, monthly intravenous immunoglobulin was started at 14 years of age. Genetics confirmed novel DOCK8 mutation exon 45 aa1970, c.5908 G > C change alanine to proline homozygous change A1970 to P1970 (Figures 1 and 2).
Figure 1:
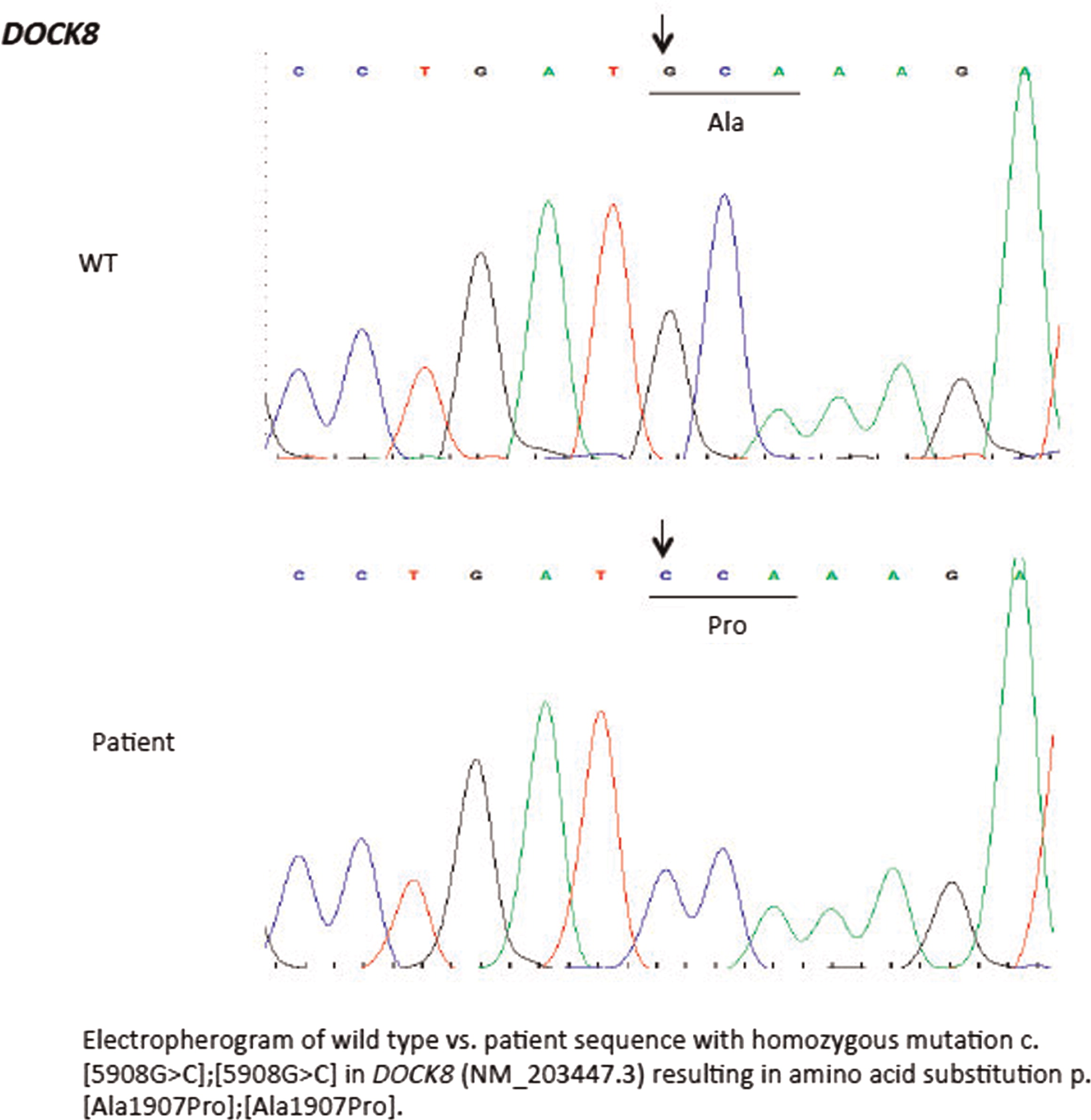
Figure 2:
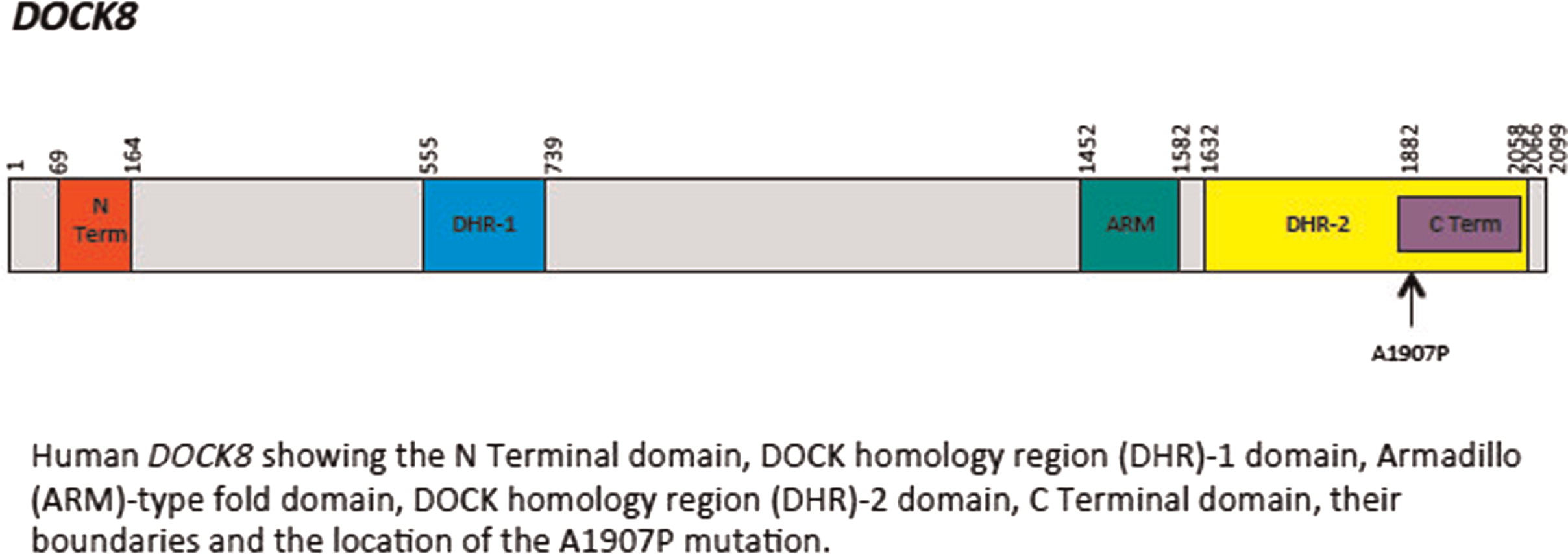
Table 1:
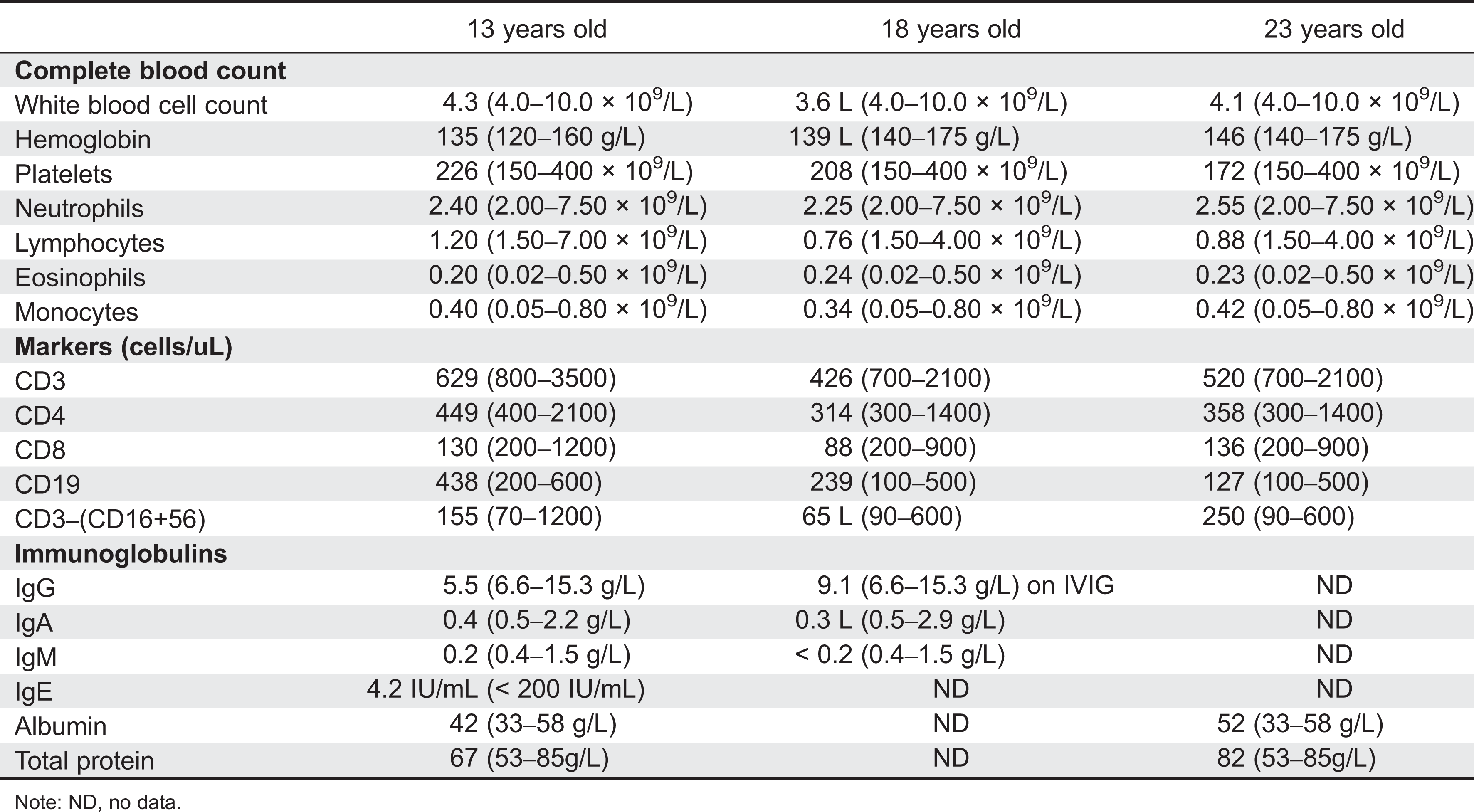
Table 2:
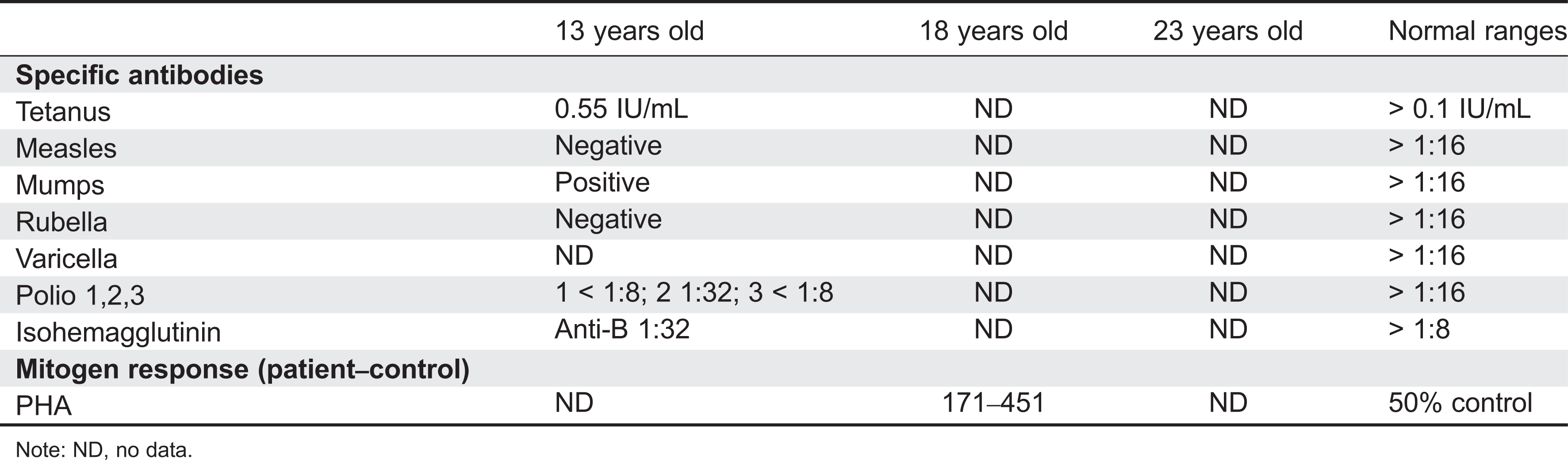
Discussion
The characteristic infectious and noninfectious features of DOCK8 mutation associated HIES are described in our patient with a novel mutation. As demonstrated in our patient, DOCK8 mutations lead to a number of cutaneous findings such as viral cutaneous infections secondary to molluscum contagiosum, herpes simplex virus (HSV), varicella-zoster virus, and human papillomavirus plus recurrent Staphylococcus aureus skin infections (Zhang et al. 2009). These infections can result in eczema herpeticum, chronic ulcerating orolabial or angogenital HSV infections, or HSV keratitis. The susceptibility to viral cutaneous infections is likely secondary to multiple factors including impaired CD8 T-cell memory response and reduced production of interferon gamma and tumor necrosis factor α by CD8+ T cells in DOCK8-deficient patients (Zhang et al. 2009; Chu et al. 2012). In addition, DOCK8 may play a role in leukocyte migration to infected skin, and the breakdown of the epidermal barrier from dermatitis can predispose patients to viral infections (Zhang et al. 2010; Chu et al. 2012). Mucocutaneous candidiasis is less common in DOCK8 patients compared with those with STAT3 mutations (Chu et al. 2012).
Similar to STAT3 mutations, DOCK8 deficiency can be difficult to differentiate from severe atopic dermatitis and other dermatitis, especially with higher rates of other allergic diseases such as food allergy also occurring with DOCK8 mutations. A recent study concluded that CD3 and CD4 T-cell lymphopenia with decreased naïve CD8 T cells and decreased memory B cells is strongly suggestive of DOCK8 deficiency rather than severe atopic dermatitis (Janssen et al. 2014b).
Sinopulmonary infections such as recurrent otitis media, sinusitis, and pneumonias are frequently observed and can be associated with complications such as bronchiectasis (Zhang et al. 2009; Alsum et al. 2013). Our patient had a past history of acute otitis media. Various pathogens have been associated with pneumonia including Streptococcus pneumonia, Haemophilus influenza, adenovirus, and respiratory syncytial virus as well as eosinophilic lung disease (Zhang et al. 2009; Tsuge et al. 2014). Recurrent sinopulmonary infections experienced in our patient and others with DOCK8 mutation are consistent with the humoral immune abnormalities identified. Opportunistic infections including Pnemocystis jiroveci and vaccine strain varicella-zoster virus induced central nervous system vasculopathy can occur, although they are less frequent (Zhang et al. 2009; Sabry et al. 2014). Diarrhea, which has been described in other patients with DOCK8 (Dinwiddie et al. 2013), was a significant complaint for our patient. Diarrhea can be secondary to infections such as salmonella, giardiasis, and norovirus (Sanal et al. 2012). Other gastrointestinal involvement includes cryptosporidium-associated liver disease (Al-Herz et al. 2012; Shah et al. 2014). Additional features described in DOCK8 patients include neurologic complications such as vasculitis, aneurysms, and central nervous system lymphoma, sclerosing cholangitis, and granulomatous soft tissue lesions (Engelhardt et al. 2009; Al-Herz et al. 2012; Sanal et al. 2012).
Unlike patients with STAT3 mutations, many patients with DOCK8 mutations also have significant allergic diseases such as food allergy causing anaphylaxis, environmental allergies, and asthma (Zhang et al. 2009). Eosinophilic esophagitis has also been reported (Zhang et al. 2009). Increased rates of allergic diseases are likely secondary to increased Th2 CD4 T cells (Lambe et al. 2011). Patients are often found to have high serum IgE and eosinophilia as the result. Although a common finding, our patient did not show any signs of allergic diseases.
Similar to many other primary immunodeficiencies, DOCK8 is associated with higher rates of autoimmunity and malignancy. In patients with long-standing cutaneous viral infections, vulvar, facial, and anal squamous cell carcinomas can occur (Zhang et al. 2009). Lymphoid malignancies such as Epstein–Barr virus (EBV) negative diffuse B-cell lymphoma, EBV-positive Burkitt lymphoma, and cutaneous T-cell lymphoma are also noted (Zhang et al. 2009). It has been suggested that impaired CD8 T cells in DOCK8 deficiency may lead to impaired tumor surveillance resulting in an increased susceptibility to malignancies (Zhang et al. 2009). Specific autoimmune conditions reported include autoimmune hemolytic anemia, colitis, vasculitis, uveitis, and systemic lupus erythematous (Al-Herz et al. 2012; Alsum et al. 2013; Jouhadi et al. 2014). It is possible that the oral ulcers and Bell's palsy experienced by our patient was an autoimmune manifestation.
Common laboratory findings include lymphopenia, eosinophilia, elevated IgE and low IgM, decreased T- and B-cell numbers, variable impaired responses to mitogen stimulation, and poor antibody response. Our patient had lymphopenia affecting mostly CD8 T cells and low immunoglobulin levels. He also had evidence of poor function low mitogen stimulation response and poor specific antibody production. He did not have eosinophilia or elevated IgE.
There is growing consensus that HSCT should be offered to patients before significant infections with associated complications and development of malignancy occur (Bittner et al. 2010; Barlogis et al. 2011; Gatz et al. 2011; McDonald et al. 2012; Metin et al. 2012). Patients with impaired antibody production may benefit from immunoglobulin replacement to reduce the frequency of sinopulmonary infections. Management of cutaneous manifestations in this patient population is difficult. Medications used to improve dermatitis such as topical corticosteroids can make cutaneous viral infections worse (Chu et al. 2012). Systemic antiviral medications are used for cutaneous viral infections although resistance has occurred, whereas topical treatments are generally not effective given the extent of the disease (Chu et al. 2012). Interferon alpha has been used with variable success for cutaneous viral infections and tumorous herpes simplex blepharoconjunctivitis (Keles et al. 2014; Papan et al. 2014). Bleach baths and topical antibacterial medications are helpful to control Staphylococcal aureus colonization. All patients require sun protection given the predisposition for skin malignancy and careful monitoring for the development of malignancy.
Overall, DOCK8 deficiency is a rare combined primary immunodeficiency characterized by recalcitrant cutaneous viral infections, recurrent sinopulmonary infections, allergic diseases, autoimmunity and malignancy with associated eosinophilia, elevated IgE, and poor number and functioning of both T and B cells. Patients with these features should be investigated for possible DOCK8 mutations. Our data presented expands the spectrum of DOCK8 mutations.
REFERENCES
Al-Herz W., Bousfiha A., Casanova J.L., Chapel H., Conley M.H., Cunningham-Rundles C., Etzioni A., Fischer A., Franco J.L., Geha R.S., Hammarström L., Nonoyama S., Notarangelo L.D., Ochs H.D., Puck J.M., Roifman C.M., Seger R., and Tang M.L. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency Front Immunol. 2011 2 54
Al-Herz W., Ragupathy R., Massaad M.J., Al-Attiyah R., Nanda A., Engelhardt K.R., Grimbacher B., Notarangelo L., Chatila T., and Geha R.S. Clinical, immunologic and genetic profiles of DOCK8-deficient patients in Kuwait Clin. Immunol. 2012 143 266 -272
Al Khatib S., Keles S., Garcia-Lloret M., Karakoc-Aydiner E., Reisli I., Artac H., Camcioglu Y., Cokugras H., Somer A., Kutukculer N., Yilmaz M., Ikinciogullari A., Yegin O., Yüksek M., Genel F., Kucukosmanoglu E., Baki A., Bahceciler N.N., Rambhatla A., Nickerson D.W., McGhee S., Barlan I.B., and Chatila T. Defects along the T[H]17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome J. Allergy Clin. Immunol. 2009 124 2 342 -348 e1 -e5
Alsum Z., Hawwari A., Alsmadi O., Al-Hissi S., Borrero E., Abu-staiteh A., Khalak H.G., Wakil S., Eldali A.M., Arnaout R., Al-ghonaium A., Al-Muhsen S., Al-Dhekri H., Al-Saud B., and Al-Mousa H. Clinical, immunological and molecular characterization of DOCK8 and DOCK8-like deficient patients: single center experience of twenty five patients J. Clin. Immunol. 2013 33 55 -67
Barlogis V., Galambrun C., Chambost H., Lamoureux-Toth S., Petit P., Stephan J.L., Michel G., Fischer A., and Picard C. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency J. Allergy Clin. Immunol. 2011 128 2 420 -422
Bittner T.C., Pannicke U., Renner E.D., Notheis G., Hoffmann F., Belohradsky B.H., Wintergerst U., Hauser M., Klein B., Schwarz K., Schmid I., and Albert M.H. Successful long-term correction of autosomal recessive hyper-IgE syndrome due to DOCK8 deficiency by hematopoietic stem cell transplantation Klin. Padiatr. 2010 222 6 351 -355
Crawford G., Enders A., Gileadi U., Stankovic S., Zhang Q., Lambe T., Crockford T.L., Lockstone H.E., Freeman A., Arkwright P.D., Smart J.M., Ma C.S., Tangye S.G., Goodnow C.C., Cerundolo V., Godfrey D.I., Su H.C., Randall K.L., and Cornal R.J. DOCK8 is critical for the survival and function of NKT cells Blood. 2013 122 2052 -2061
Chu E., Freeman A., Jing H., Cowen E., Davis J., Su H., Holland S.M., and Chanco Turner M.L. Cutaneous manifestations of DOCK8 deficiency syndrome Arch. Dermatol. 2012 148 1 79 -84
Dasouki M., Okonkwo K.C., Ray A., Folmsbeel C.K., Gozales D., Keles S., Puck J.M., and Chatila T. Deficient T cell receptor excision circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: implications for pathogenesis and potential detection by newborn screening Clin. Immunol. 2011 141 2 128 -132
Dinwiddie D.L., Kingsmore S.F., Caracciolo S., Rossi G., Moratto D., Mazza C., Sabelli C., Bacchetta R., Passerini L., Magri C., Bell C.J., Miller N.A., Hateley S.L., Saunders C.J., Zhang L., Schroth J.P., Barlati S., and Badolato R. Combined DOCK8 and CLEC7A mutations causing immunodeficiency in 3 brothers with diarrhea, eczema, and infections J. Allergy Clin. Immunol. 2013 131 2 594 -597
Engelhardt K.R., McGhee S., Winkler S., Sassi A., Woellner C., Lopez-Herrera G., Chen A., Kim H.S., Lloret M.G., Schulze I., Ehl S., Thiel J., Pfeifer D., Veelken H., Niehues T., Siepermann K., Weinspach S., Reisli I., Keles S., Genel F., Kutukculer N., Camcioğlu Y., Somer A., Karakoc-Aydiner E., Barlan I., Gennery A., Metin A., Degerliyurt A., Pietrogrande M.C., Yeganeh M., Baz Z., Al-Tamemi S., Klein C., Puck J.M., Holland S.M., McCabe E.R., Grimbacher B., and Chatila T.A. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal recessive for of hyper-IgE syndrome J. Allergy Clin. Immunol. 2009 124 6 1289 -1302
Gatz S.A., Benninghoff U., Schulz C., Schulz A., Honig M., Pannicke U., Holzmann K.H., Schwarz K., and Friedrich W. Curative treatment of autosomal-recessive hyper-IgE syndrome by hematopoietic cell transplantation Bone Marrow Transpl. 2011 46 552 -556
Holland S.M., DeLeo F.R., Elloumi H.Z., Hsu A.P., Uzel G., Brodsky N., Freeman A.F., Demidowich A., Davis J., Turner M.L., Anderson V.L., Darnell D.N., Welch P.A., Kuhns D.B., Frucht D.M., Malech H.L., Gallin J.I., Kobayashi S.D., Whitney A.R., Voyich J.M., Musser J.M., Woellner C., Schäffer A.A., Puck J.M., and Grimbacher B. STAT3 mutations in the hyper-IgE syndrome N Engl J Med. 2007 357 16 1608 -1619
Jabara H.H., McDonald D.R., Janssen E., Massaad M.J., Ramesh N., Borzutzky A., Rauter I., Benson H., Schneider L., Baxi S., Recher M., Notarangelo L.D., Wakim R., Dbaibo G., Dasouki M., Al-Herz W., Barlan I., Baris S., Kutukculer N., Ochs H.D., Plebani A., Kanariou M., Lefranc G., Reisli I., Fitzgerald K.A., Golenbock D., Manis J., Keles S., Ceja R., Chatila T.A., and Geha R.S. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation Nat. Immunol. 2012 13 6 612 -620
Janssen E., Morbach H., Ullas S., Bannock J.M., Massad C., Menard L., Barlan I., Lefranc G., Su H., Dasouki M., Al-Herz W., Keles S., Chatila T., Geha R., and Meffre E. Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells J. Allergy Clin. Immunol. 2014a Article in press
Janssen E., Tsitsikov E., Al-Herz W., Lefranc G., Megarbane A., Dasouki M., Bonilla F.A., Chatila T., Schneider L., and Geha R. Flow cytometry biomarkers distinguish DOCK8 deficiency from severe atopic dermatitis Clin. Immunol. 2014b 150 220 -224
Jouhadi Z., Khadir K., Ailal F., Bouayad K., Nadifi S., Engelhardt K., and Grimbacher B. Ten-year follow-up of a DOCK8-deficient child with features of systemic lupus erythematous Pediatrics. 2014 134 5 e1458 -e1463
Keles S., Jabara H.H., Reisli I., McDonald D.R., Barlan I., Hanna-Wakim R., Dbaibo G., Lefranc G., Al-Herz W., Geha R.S., and Chatila T.A. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with IFN-a 2b therapy J. Allergy Clin. Immunol. 2014 133 6 1753 -1755.e3
Lambe T., Crawford G., Johnson A.L., Crockford T.L., Bouriez-Jones T., and Smyth A.M. DOCK8 is essential for T-cell survival and the maintenance of CD8+ T-cell memory Eur. J. Immunol. 2011 41 12 3423 -3435
McDonald D.R., Massaad M.J., Johnston A., Keles S., Chatila T., Geha R.S., and Pai S.Y. Successful engraftment of donor marrow after allogeneic hematopoietic cell transplantation in autosomal-recessive hyper-IgE syndrome caused by dedicator of cytokinesis 8 deficiency J. Allergy Clin. Immunol. 2012 126 6 1304 -1305
Meller N., Merlot S., and Guda C. CZH proteins: a new family of Rho-GEFs J. Cell. Sci. 2005 118 4937 -4946
Metin A., Tavil B., Azık F., Azkur1 D., Ok-Bozkaya I., Kocabas C., Tunc B., and Uckan D. Successful bone marrow transplantation for DOCK8 deficient hyper IgE syndrome Pediatr. Transpl. 2012 16 4 398 -399
Minegishi Y., Saito M., Morio T., Watanabe K., Agematsu K., Tsuchiya S., Takada H., Hara T., Kawamura N., Ariga T., Kaneko H., Kondo N., Tsuge I., Yachie A., Sakiyama Y., Iwata T., Bessho F., Ohishi T., Joh K., Imai K., Kogawa K., Shinohara M., Fujieda M., Wakiguchi H., Pasic S., Abinun M., Ochs H.D., Renner E.D., Jansson A., Belohradsky B.H., Metin A., Shimizu N., Mizutani S., Miyawaki T., Nonoyama S., and Karasuyama H. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity Immunity. 2006 25 5 745 -755
Minegishi Y., Saito M., Tsuchiya S., Tsuge I., Takada H., Hara T., Kawamura N., Ariga T., Pasic S., Stojkovic O., Metin A., and Karasuyama H. Dominant negative mutations in the DNA binding domain of STAT3 cause hyper IgE syndrome Nature. 2007 448 1058 -62
Papan C., Hagl B., Heinz V., Albert M.H., Ehrt O., Sawalle-Belohradskya J., Neumann J., Ries M., Bufler P., Wollenberg A., and Renner E.D. Beneficial IFN-a treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency J. Allergy Clin. Immunol. 2014 133 5 1456 -1458
Randall K., Lambe T., Johnson A., Treanor B., Kucharska E., Domaschenz H., Whittle B., Tze L., Enders A., Crockford T., Bouriez-Jones T., Alston D., Cyster J., Lenardo M., Mackay F., Deenick E., Tangye S., Chan T., Camidge T., Brink R., Vinuesa C., Batista F., Cornall3 R., and Goodnow C. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production Nat. Immunol. 2009 10 12 1283 -1291
Ruusala A. and Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology FEBS Lett. 2004 572 1–3 159 -166
Sabry A., Hauk P.J., Jing H., Su H.C., Stence N.V., Mirsky D.M., Nagel M.A., Abbott J.K., Dragone L.L., Armstrong-Wells J., and Curtis D.J. Vaccine strain varicella-zoster virus–induced central nervous system vasculopathy as the presenting feature of DOCK8 deficiency J. Allergy Clin. Immunol. 2014 133 4 1225 -1227
Sanal O., Jing H., Ozgur T., Ayvaz D., Strauss-Albee D.M., Ersoy-Evans S., Tezcan I., Turkkani G., Matthews H.F., Haliloglu G., Yuce A., Yalcin B., Gokoz O., Oguz K.K., and Su H.C. Additional diverse findings expand the clinical presentation of DOCK8 deficiency J. Clin. Immunol. 2012 32 698 -708
Shah T., Hadzic N., and Jones A. Dedicator of cytokinesis 8 deficiency: a predisposition to sclerosing cholangitis Clin. Immunol. 2014 155 1 71 -73
Su H.C. DOCK8 (Dedicator of cytokinesis 8) deficiency Curr. Opin. Allergy Clin. Immunol. 2010 10 6 515 -520
Tsuge I., Ito K., Ohye T., Kando N., Kondo Y., Nakajima Y., Inuo C., Kurahashi H., and Urisu A. Acute eosinophilic pneumonia occurring in dedicator of cytokinesis 8 (DOCK8) deficient patient Ped. Pulmonol. 2014 49 3 E52 -E55
Xue, L., Yang, Y., and Wang, S. 2014. A novel large deletion of the DOCK8 gene in a Chinese family with autosomal-recessive hyper-IgE syndrome. JEADV.
Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G., Holland S.M., and Su H.C. Combined immunodeficiency associated with DOCK8 mutations N. Engl. J. Med. 2009 361 21 2046 -2055
Zhang Q., Davis J.C., Dove C.G., and Su H.C. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome Dis. Mark. 2010 29 3–4 131 -139
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: 39 - 46
History
Received: 8 December 2014
Accepted: 11 December 2014
Accepted manuscript online: 11 December 2014
Version of record online: 11 December 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Alison Haynes. 2015. Novel DOCK8 Mutation. LymphoSign Journal.
2(1): 39-46. https://doi.org/10.14785/lpsn-2014-0023
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
