Cardiovascular abnormalities in primary immunodeficiency diseases
Abstract
In recent years, increasing numbers of patients with primary immune deficiency (PID) are being recognized as also suffering from cardiovascular system (CVS) abnormalities. These CVS defects might be explained by infectious or autoimmune etiologies, as well as by the role of specific genes and the immune system in the development and function of CVS tissues. Here, we provide the first comprehensive review of the clinical, potentially pathogenic mechanisms, and the management of PID, as well as the associated immune and CVS defects. In addition to some well-known associations of PID with CVS abnormalities, such as DiGeorge syndrome and CHARGE anomaly, we describe the cardiac defects associated with Omenn syndrome, calcium channel deficiencies, DNA repair defects, common variable immunodeficiency, Roifman syndrome, various neutrophil/macrophage defects, FADD deficiency, and HOIL1 deficiency. Moreover, we detail the vascular abnormalities recognized in chronic mucocutaneous candidiasis, chronic granulomatous disease, Wiskott–Aldrich syndrome, Schimke immuno-osseus dysplasia, hyper-IgE syndrome, MonoMAC syndrome, and X-linked lymphoproliferative disease. In conclusion, the expanding spectrum of PID requires increased alertness to the possibility of CVS involvement as an important contributor to the diagnosis and management of these patients.
Introduction
Primary immunodeficiency diseases (PID) encompass a broad spectrum of inheritable disorders that are commonly associated with increased susceptibility to infections, immune dysregulation, and malignancies. Early recognition and correction of the immune deficiency is crucial. Patients with profound T-cell immune deficiency such as severe combined immunodeficiency (SCID) should be treated by allogeneic hematopoietic stem cell transplantation (HSCT) or autologous gene therapy, whereas antibody deficiency can be corrected by immunoglobulin replacement.
The important role of the immune system in the development and function of many organs as well as the effects of gene defects in nonimmune tissues contributes to the multi-system abnormalities often observed in patients suffering from PID. Additionally, the nonimmune abnormalities can be a clue to establishing the specific etiology of the PID, anticipating potential complications or choosing best management options. Indeed, nonimmune neurological, bone, gastrointestinal, skin, and endocrine abnormalities are often associated with various PID, such as purine nucleoside phosphorylase deficiency, adenosine deaminase deficiency, cartilage–hair hypoplasia, ectodermal dysplasia, etc. In recent years, particularly with the significant increase in PID for which molecular defects are identified, many PID conditions are recognized to be associated with cardiovascular system (CVS) abnormalities.
Here we provide a comprehensive review of PID associated with CVS defects. The review was performed using the PubMed database. The International Union of Immunological Societies Expert Committee for Primary Immunodeficiency classification system (Bousfiha et al. 2013) was used to systematically search for articles reporting PID associated with cardiac or vascular malformations. Additional associations were identified through an open PubMed search by combining terms such as “primary immunodeficiency” and “cardiac” or “heart” or “vascular”. CVS complications secondary to infectious etiologies as well as disorders caused predominantly or typically by rheumatologic and autoimmune mechanisms were excluded.
The review is intended for both cardiologists and immunologists. Therefore, following a brief description of the clinical features and pathogenesis of the specific condition, the immunological and CVS abnormalities are provided as well as management strategies.
Also, as a useful reference, we included 3 Appendices that summarize the cardiac and vascular abnormalities associated with PID (Appendix A1), the PID associated with cardiac abnormalities (Appendix A2), and the PID associated with vascular abnormalities (Appendix A3). Additionally, Appendix B1 summarizes the acronyms used in this review.
Predominantly T lineage defects
Omenn syndrome
Description: Omenn syndrome (OMIM# 603554), a combination of generalized severe erythroderma, lymphadenopathy, eosinophilia, and profound immunodeficiency in infancy, was first described in 1965. Other manifestations include hepatosplenomegaly, failure to thrive, diarrhea, and alopecia. Laboratory investigations often reveal eosinophilia and elevated IgE (Villa et al. 2008).
Pathogenesis: Omenn syndrome can occur with practically all profound T-cell immune deficiencies, such as those caused by defects in recombinant activating genes (RAG) 1 and 2, ARTEMIS (DCLRE1C gene), the RNA component of mitochondrial RNA processing endoribonuclease gene (RMRP), DNA Ligase IV, interleukin 7 receptor α, common γ chain, CD3δ, adenosine deaminase, and purine nucleoside phosphorylase deficiencies, DiGeorge syndrome (DGS), etc. (Villa et al. 1998, 2008; Ege et al. 2005; Zhang et al. 2005; Grunebaum et al. 2008). Common to these conditions is the ability of some T cells to mature in the thymus, escape proper selection, and expand in the periphery (Aleman et al. 2001; Poliani et al. 2009).
Immune abnormalities: Patients with Omenn syndrome have severely impaired cellular and humoral immunity; however, in contrast to typical severe combined immunodeficiency, T-cell numbers may be normal or increased. Proliferation of T cells ex vivo in response to various stimuli is often depressed, but might be present. T-cell receptor excision circles (TREC), which represent new thymus emigrants, are markedly reduced (Roifman et al. 2012). Significantly limited T-cell diversity and T-cell clonal expansion, increased IgE, and eosinophils are additional hallmarks of Omenn syndrome (Somech et al. 2009).
CVS abnormalities: Cardiac manifestations of Omenn syndrome have rarely been described. An infant with Omenn syndrome was found to have biventricular hypertrophy, impaired left ventricular systolic function, and severe sinus bradycardia, possibly secondary to endomyocardial disease caused by eosinophilia (Bruckmann et al. 1991).
Treatment: Antimicrobial and anti-inflammatory medications such as systemic steroids or cyclosporine A together with strict isolation can provide temporary benefit. Ultimately, Omenn syndrome can only be cured by HSCT, preferably from a human leucocyte antigen (HLA)-matched sibling or unrelated donor (Nahum et al. 2009).
Calcium channel deficiency
Description: ORAI-1 deficiency (OMIM# 610277) is an autosomal recessive form of SCID, characterized by recurrent infections in infancy, congenital muscular hypotonia, and anhydrotic ectodermal dysplasia (Feske et al. 2010). It is also associated with autoimmunity, developmental delay, and failure to thrive (McCarl et al. 2009).
Pathogenesis: This clinical syndrome is caused by a mutation in the ORAI1 gene, encoding part of the calcium release activated calcium channel. T cells rely on this channel for calcium influx for subsequent activation. Thus, mutations in ORAI1 lead to severely impaired T-cell function. Calcium influx secondary to calcium release activated calcium channel function is also crucial for skeletal muscle function and (or) differentiation, leading to the congenital myopathy (McCarl et al. 2009; Feske et al. 2010). Ectodermal dysplasia is characterized by an inability to sweat and impaired thermoregulation, for which the precise mechanism is not clear.
Immune abnormalities: ORAI-1 deficient patients suffer from recurrent viral (particularly herpes viruses), bacterial, and fungal infections despite having normal T-cell and B-cell numbers. Lymphocyte development is unaffected; however, T-cell activation as well as the functions of other immune cells, including B cells, are severely impaired. Natural killer (NK) cell functions, including degranulation, cytokine production, and cytolytic activity may also be impaired (Notarangelo 2013).
CVS abnormalities: CVS abnormalities have not been reported in ORAI-1 deficient patients; however, inactivation of ORAI1 in Zebra fish causes reduced ventricular systolic function, bradycardia, and heart failure (Volkers et al. 2012), suggesting that cardiomyopathy may co-exist with the skeletal myopathy.
A similar, albeit less severe, immunodeficiency is present among patients with STIM1 mutations (OMIM# 605921) that also disrupt calcium entry; however, cardiac defects are not reported (Feske et al. 2010).
Treatment: HSCT can correct the immunodeficiency; however, the skin and muscle abnormalities persist.
Chronic mucocutaneous candidiasis
Description: Chronic mucocutaneous candidiasis (CMCC) is a group of disorders characterized by recurrent Candida infections of the skin, nails, and mucous membranes, which may appear in infancy or later in life and are often accompanied by autoimmune endocrine abnormalities such as hypo-parathyroidism and adrenal insufficiency. Indeed, some patients are categorized as suffering from autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy or autoimmune polyendocrine syndrome type 1. Patients may also develop chronic keratitis, eczema, and other autoimmune phenomena (Liu et al. 2011).
Pathogenesis: CMCC phenotype is associated with mutations in several genes. An autosomal recessive variant of CMCC is caused by defects in the autoimmune regulator (AIRE) gene (OMIM# 240300) encoding a transcription factor expressed in the medulla of the thymus and is thought to be important for presentation of tissue-specific self-antigens to maturing thymocytes. DECTIN-1 and CARD9 deficiency have also been associated with autosomal recessive CMCC. Mutations in STAT1, a transcription factor involved in multiple signaling pathways, are the main cause of autosomal dominant CMCC (OMIM# 600555), although the precise mechanism for the susceptibility to candidiasis is still not clear (Liu et al. 2011).
Immune abnormalities: Immune dysfunction in CMCC is variable. Typically, Candida infections are limited to the mucocutaneous membranes and are not invasive, although patients may display T-cell deficiency, manifesting with increased susceptibility to cytomegalovirus and other viruses (Nahum et al. 2012) as well as disseminated coccidioidomycosis and histoplasmosis (Sampaio et al. 2013). Some patients do not produce appropriate antibodies, resulting in increased frequency of sinopulmonary infections. Laboratory findings in patients with CMCC include normal or reduced in vitro and in vivo T-cell responses to Candida antigens, elevated IgE, and detection of various auto-antibodies (Kirkpatrick, 2001; Puel et al. 2010). In addition to glandular damage, there have been reports of autoimmune cytopenias, hepatitis, colitis, etc. (Hori et al. 2012). Some patients might also develop thymoma or hemophagocytic lymphohistiocytosis or present as Omenn syndrome (Cavadini et al. 2005).
CVS abnormalities: CMCC is associated with vasculitis, particularly cerebral vasculitis, with severe neurologic complications (Grouhi et al. 1998; Nagashima et al. 2000). A brain biopsy in one patient demonstrated periarterial mononuclear cell infiltrates, which is suggestive of endarteritis (Grouhi et al. 1998). Subsequently, patients with CMCC with dominant STAT1 mutations were noted to suffer from cerebral aneurysms and infarctions (Marazzi et al. 2008; Uzel et al. 2013). It is still not clear whether these cerebral abnormalities are caused by unrecognized infections, autoimmune phenomena, or structural defect directly related to either the gene defect or the potential involvement of other tissues.
Treatment: Oral and topical antifungal medications are the mainstay of treatment for CMCC (Kirkpatrick 2001). Immunoglobulin replacement has been beneficial for patients with antibody deficiency, whereas immune suppressive medications have been used for the autoimmune phenomena. The benefit of HSCT for the very severe forms of CMCC is still not established.
STK-4 deficiency
Description: Autosomal recessive mutations in STK4, also known as MST1 (OMIM# 614868), were found among consanguineous Iranian and Turkish families with lymphopenia, intermittent neutropenia, and structural cardiac defects (Abdollahpour et al. 2012; Nehme et al. 2012).
Pathogenesis: Increased apoptosis of STK-4 deficient T cells and granulocytes may explain the reduced numbers of cells.
Immune abnormalities: Patients suffer from recurrent bacterial infections, including sinusitis, pneumonia, and meningitis, as well as herpes and papilloma virus infections, mucocutaneous candidiasis, cutaneous warts, and skin abscesses. Patients may present in infancy or childhood. Dermatitis and autoimmunity, including hemolytic anemia, also develop in some patients. Immune evaluations demonstrate lymphopenia with paucity of T cells, particularly CD4+ cells. Proliferation in response to stimulation with mitogens or antigens is difficult to assess because of the rapid death of STK-4 deficient T cells in vitro. B-cell numbers are reduced; however, IgG and IgE levels are increased and production of various antibodies is appropriate. The neutropenia might be intermittent in some patients. Contrary to many other forms of congenital neutropenia, the bone marrow of the patients with STK-4 deficiency appears normal. Epstein–Barr virus associated lymphadenopathy, phenotypically resembling lymphoplasmacytic lymphoma can also occur (Abdollahpour et al. 2012).
CVS abnormalities: Atrial septal defect type II, patent foramen ovale as well as mitral, tricuspid, and pulmonary insufficiency can be identified in some patients. Interestingly, STK-4 deficient mice develop dilated cardiomyopathy secondary to increased apoptosis of myocytes (Yamamoto et al. 2003).
Treatment: Antimicrobial and antiviral prophylaxis as well as immunoglobulin infusions may help prevent the frequent infections in STK-4 deficient patients. Close monitoring for lymphoproliferative disease and cardiac defects are warranted. HSCT appears to cure the immunodeficiency (Nehme et al. 2012).
Lymphocyte-specific protein tyrosine kinase deficiency
A single patient with homozygous lymphocyte-specific protein tyrosine kinase (LCK) mutation (OMIM# 153390) who suffered from recurrent infections and retinal vasculitis was identified by Hauck et al. (2012).
Pathophysiology: Despite the important role of LCK in T cells, some T cells do develop and escape the thymus, possibly contributing to the immune dysregulation observed in LCK deficiency.
Immune abnormalities: The patient suffered from recurrent respiratory tract infections, protracted gut disease, failure to thrive, nodular skin lesions, arthritis, and autoimmune thrombocytopenia from an early age. Immune evaluations revealed severe CD4+ T-cell lymphopenia with oligoclonal TCRαβ+ T cells, increase in central memory CD4+ cell, and “exhausted” T-effector memory CD8+ cells. Activation of T cells with anti-CD3 demonstrated markedly reduced early tyrosine phosphorylation events and abrogated Ca2+ mobilization resulting in a severe proliferation defect. IgM levels were increased, whereas antibodies to vaccines were low. Autoantibodies to multiple self-antigens were present. The low number of Treg cells and the reduced activation-induced cell death of the patient's T cells might have contributed to the immune dysregulation (Hauck et al. 2012).
CVS abnormalities: The patient suffered from pericarditis, possibly related to capillary leak syndrome as well as retinal vasculitis (Hauck et al. 2012).
Treatment: Antibiotic prophylaxis and immunoglobulin replacement are warranted. This single patient with LCK deficiency died shortly after HSCT from veno-occlusive disease.
DNA repair defects
Ataxia–telangiectasia
Description: Ataxia–telangiectasia (AT) is a well-described autosomal recessive disorder characterized by cerebellar ataxia, oculomotor apraxia, telangiectasia, and immunodeficiency (OMIM# 208900). Patients’ cells are also highly sensitive to irradiation. Progressive ataxia is often noted at 2–3 years of age. Other neurologic features include nystagmus, dysarthria, and choreoathetosis. Telangiectasia in the eyes and skin typically follow the neurological symptoms. Patients are at significant risk for malignancy. Elevated α-fetoprotein blood levels are very helpful in establishing the diagnosis of AT beyond the first few months of life, although other genetic defects such as ataxia with oculomotor apraxia type 1 and type 2 can also cause ataxia and increased α-fetoprotein (Nanetti et al. 2013).
Pathogenesis: AT is caused by defects in the AT mutated protein, a kinase with significant role in sensing and repairing double-stranded DNA breaks. Accordingly, AT mutated protein defects lead to abnormal DNA repair, resulting in accumulation of damaged genetic material in neuronal and other cells (Perlman et al. 2012).
Immune abnormalities: The immunodeficiency is variable. Sinopulmonary infections are frequent. Typical laboratory manifestations include decreased IgA, IgE, and IgG subclasses and antibody responses, whereas IgM might be elevated, resulting in a phenotype similar to that of hyper-IgM syndrome. Lymphopenia, depressed T-cell function, and reduced TREC can also be found, suggesting that some patients might be diagnosed through SCID newborn screening.
CVS abnormalities: Mitral valve prolapse, mitral regurgitation, and tricuspid regurgitation are found in few patients with AT (Bastianon et al. 1993). Interestingly, carriers of mutations in a single allele AT mutated kinase are more susceptible to ischemic heart disease, from an as yet unknown cause (Su and Swift 2000).
Treatment: Currently, there is no cure for AT. Patients should avoid exposure to ionizing radiation. Physical, occupational, and speech therapy, as well as a healthy diet and lifestyle can improve quality of life (Perlman et al. 2012). Antibiotic prophylaxis or immunoglobulin therapy can aid patients with recurrent infections or documented evidence of cellular and humoral immune deficiency.
Nijmegen breakage syndrome
Description: Nijmegen breakage syndrome (OMIM# 251260) is a rare autosomal recessive disorder characterized by chromosomal instability. Common clinical features include microcephaly, immunodeficiency, radiation hypersensitivity, and predisposition to malignancy. Patients with Nijmegen breakage syndrome may also display typical bird-like facial features with a prominent mid-face, micrognathia, and beaked nose. Nijmegen breakage syndrome is also associated with growth delay and premature ovarian failure in females (Chrzanowska et al. 2012).
Pathogenesis: Nijmegen breakage syndrome is caused by mutations in the NBS1 gene, encoding the Nibrin protein. Nibrin is a component of the MRN (MRE11–RAD50–NBN) complex that is critical to the DNA damage response system, along with the AT mutated protein. Thus, defective Nibrin leads to impaired DNA repair and a phenotype similar to AT (Chrzanowska et al. 2012).
Immune abnormalities: There are significant variations in the immunodeficiency among Nijmegen breakage syndrome patients. Most frequently, patients have reduced immunoglobulin levels. B and T cells may also be decreased (Chrzanowska et al. 2012).
CVS abnormalities: One patient with Nijmegen breakage syndrome was reported to suffer from a large sub-aortic ventricular septum defect (Tekin et al. 2002), possibly related to the abundant expression of NBS1 in the cardiac muscle, as demonstrated in mouse embryos (Wilda et al. 2000).
Treatment: Antibiotic prophylaxis and immunoglobulin replacement therapy may prevent infections, while physiotherapy and occupational therapy may help improve quality of life. As in AT, avoiding exposure to ionizing radiation and regular screening for malignancy is very important (Chrzanowska et al. 2012). Recently, favorable outcome following HSCT was reported for few patients with Nijmegen breakage syndrome with severe immunodeficiency or malignancy (Albert et al. 2010).
Predominantly antibody deficiencies
Common variable immunodeficiency
Description: Common variable immunodeficiency (CVID) is a heterogeneous group of disorders characterized by hypogammaglobulinemia, abnormal production of antibodies, and no other known cause for the immune defect. Infections typically occur in early- to mid-adulthood but can present in childhood or even among aging individuals. Few CVIDs are secondary to single gene defects that interfere with B-cell development or function, yet the majority of cases have no underlying cause (Cunningham-Rundles 2012).
Immune abnormalities: Most patients suffer from recurrent sinopulmonary infections, as well as arthritis, osteomyelitis, meningitis, or sepsis caused by bacterial and viral pathogens. Many patients come to medical attention due to inflammatory or autoimmune complications (Cunningham-Rundles 2012). The latter include organ-specific and hematological autoimmune cytopenias, lymphocyte infiltration into the lung and gastrointestinal tract, lymphadenopathy, splenomegaly, and granulomatous disease (Conley et al. 2009). Typically, IgG is reduced, whereas IgA and IgM may be normal, elevated, or reduced. It is important to exclude secondary causes for low IgG, such as gastrointestinal or urine loss, medication effects, malignancy, etc. Specific antibodies to infections or vaccinations are typically absent. Autoantibodies may appear with or without clinical autoimmune manifestations. The number of circulating B cells is usually normal (Conley et al. 2009) while kappa-deleting recombination excision circles are reduced in some patients (Kamae et al. 2013).
CVS abnormalities: CVID is not classically associated with CVS disease; however, few patients with CVID have aortic root dilatation (Johnston et al. 2004). A 16-year-old boy with CVID was reported to have an aortic aneurysm extending to the renal arteries (Kashef et al. 2011), whereas giant cell myocarditis, a rare aggressive form of autoimmune myocarditis, was found in a teenager with CVID (Laufs et al. 2002). Lastly, a 41-year-old woman, diagnosed with CVID later in life was found to have an asymptomatic right-sided aortic arch (Yalcin et al. 2004).
Treatment: The mainstay treatment for primary B-cell defects, including CVID, is immunoglobulin replacement. Patients should also receive prompt and appropriate antibiotic therapy for infections. Antibiotic prophylaxis against respiratory infections might also be beneficial (Hoernes et al. 2011).
TWEAK deficiency
Autosomal-dominant deficiency in tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) can cause recurrent infections and impaired antibody responses to protein and polysaccharide vaccines (Wang et al. 2013). Patients may also develop chronic thrombocytopenia, intermittent neutropenia, and multiple warts. Patients do not suffer from CVS abnormalities, yet TWEAK, a member of the TNF superfamily ligands, has a direct effect on cardiomyocytes (Jain et al. 2009).
Pathogenesis: The immune abnormalities are attributed to inhibition of B-cell activating factor signaling in B cells secondary to impaired activation of the NF-κB and MAPK pathways. TWEAK action on the cardiac Fn14 receptor is thought to cause cardiomyocyte elongation and cardiac fibrosis.
Immune abnormalities: Patients with TWEAK deficiency suffer from recurrent sinopulmonary infections from infancy, as well as meningitis and osteomyelitis. The frequency of infections may decrease with age. Immune evaluations demonstrate low to normal IgG levels as well as low levels of IgM and IgA. Antibody responses to T-cell dependent and polysaccharide antigens are absent. Nearly all of the B cells are of naive phenotype.
CVS abnormalities: Elevated circulating TWEAK levels were found among patients with idiopathic dilated cardiomyopathy (Jain et al. 2009).
Treatment: Antibiotic and immunoglobulin treatment and prophylaxis may help the infections. Careful evaluation for the possibility of developing CVS abnormalities is needed.
Predominantly defects of neutrophils/macrophages number and function
Severe congenital neutropenia type 4/G6PC3 deficiency
Description: Severe congenital neutropenia (SCN) type 4 (OMIM# 612541) is a rare autosomal recessive disorder characterized by neutropenia, recurrent bacterial infections, and increased risk of leukemia, structural heart defects, and urogenital malformations (Boztug et al. 2009). Prominent superficial venous pattern, myopathy, increased skin laxity, congenital hydronephrosis, and thrombocytopenia, which might be intermittent, are observed in some patients (Boztug et al. 2009; Banka et al. 2011).
Pathogenesis: SCN4 is caused by mutations in the glucose-6-phosphatase, catalytic subunit 3 (G6PC3) gene, resulting in decreased function of the glucose-6-phosphatase enzyme and possibly decreased intra-cellular glucose concentrations (Boztug et al. 2009; Banka et al. 2011). G6PC3 deficiency leads to increased neutrophil apoptosis. Bone marrow examination of some patients with G6PC3 deficiency demonstrates maturation arrest of neutrophils, whereas in other patients myelokathexis is evident. Enhanced apoptosis may also be responsible for the congenital malformations associated with SCN4 (Boztug et al. 2009; Banka et al. 2011).
Immune abnormalities: Neonatal sepsis, frequent sinopulmonary infections, oral ulcers, etc., are attributed to the severe neutropenia. One patient was also recently described with T-cell lymphopenia (Begin et al. 2013).
CVS abnormalities: SCN4 is associated with structural heart abnormalities including atrial septum defects, patent ductus arteriosus, mitral regurgitation, anomalous pulmonary veins, pulmonary stenosis, and cor triatriatum (Boztug et al. 2009; Banka et al. 2011).
Treatment: Neutrophil counts often improve following granulocyte cell stimulating factor administration (McDermott et al. 2010). Cardiac and (or) urogenital malformations may need surgical intervention.
Barth syndrome
Description: Barth syndrome (OMIM# 302060) is an X-linked disorder characterized by neutropenia, cardiac disease, skeletal myopathy, developmental delay, and failure to thrive. Organic aciduria and hypocholesterolemia may also be present. The phenotype varies in severity among patients and may change over time (Schlame and Ren 2006; Jefferies 2013).
Pathogenesis: Barth syndrome is caused by a mutation in the TAFAZZIN gene on chromosome Xq28. This results in impaired phospholipid metabolism and disruption of mitochondrial biogenesis. Muscle and heart tissue have high-energy turnover and require strict structural organization of mitochondria. Thus, the abnormal mitochondria might lead to the skeletal and cardiac myopathy. Abnormal mitochondria also cause exposure of phosphatidylserine in cell membranes, leading to increased clearance of neutrophils by tissue macrophages (Schlame and Ren 2006; Finsterer and Frank 2013).
Immune abnormalities: Neutropenia is the main immunological feature. In some cases, it is intermittent or not present at all (Spencer et al. 2006).
CVS abnormalities: Dilated cardiomyopathy is the classic cardiac feature associated with Barth syndrome. Less common manifestations include hypertrophic cardiomyopathy, left ventricular noncompaction, endocardial fibroelastosis, ventricular arrhythmias, and thromboembolic cerebrovascular events (Jefferies 2013).
Treatment: There is no cure for Barth syndrome. Supportive therapy includes physiotherapy, antibiotics for any bacterial infections, heart function monitoring, and medications if necessary. In severe cases, heart transplant might be required (Spencer et al. 2006).
Cohen syndrome
Description: Cohen syndrome (OMIM# 216550) is an autosomal recessive disorder characterized by developmental delay, microcephaly, typical facial features, ophthalmologic abnormalities, and granulocytopenia. Facial features include high-arched or wave-like eyelids, short philtrum, and thick hair with a low hairline. Cohen syndrome patients often develop retinochoroidal dystrophy and myopia by 5 years of age. Other manifestations include joint laxity, narrow hands and feet, sandal-gap toes, and a sociable disposition (Kivitie-Kallio and Norio 2001).
Pathogenesis: Cohen syndrome is caused by mutations in the COH1 gene on chromosome 8 that might be involved in vesicle-mediated sorting and intracellular protein transport (Kolehmainen et al. 2003).
Immune abnormalities: The neutropenia in Cohen syndrome, thought to be secondary to a defect in myeloid differentiation, is typically intermittent and usually does not predispose patients to infections (Kivitie-Kallio and Norio 2001).
CVS abnormalities: Some patients suffer from mitral valve prolapse and decreasing left ventricle function with age (Sack and Friedman 1980; Mehes et al. 1988; Kivitie-Kallio et al. 1999).
Treatment: The retinopathy requires regular monitoring and eyeglasses. The neutropenia does not require any specific treatment (Kivitie-Kallio and Norio 2001).
Shwachman–Diamond syndrome
Description: Shwachman–Diamond syndrome (SDS) is an autosomal recessive disorder (OMIM# 260400) characterized by neutropenia, severe exocrine pancreatic insufficiency, liver abnormalities, and bone marrow failure or myelodysplastic syndrome. Additionally, patients are predisposed to develop leukemia (Dror et al. 2011). SDS is often associated with neuro-developmental abnormalities and metaphyseal dysplasia.
Pathogenesis: SDS is caused by mutations in the SBDS gene on chromosome 7. Although the precise function of the protein encoded by the SBDS gene is unknown, it is thought to be involved in ribosome synthesis (Sen et al. 2011).
Immune abnormalities: SDS patients are prone to recurrent bacterial and viral infections, especially otitis media, pneumonia, septicemia, osteomyelitis, and skin infections. The neutropenia usually presents at an early age and can be either intermittent or chronic. Some patients with SDS have variable immune abnormalities (Dror et al. 2001). These include low IgG or IgG subclasses, low percentage of circulating B cells, decreased in vitro B-cell proliferation, and a lack of specific antibody production. T lineage abnormalities among these patients comprise a low percentage of total circulating T cells or CD3+/CD4+ T-cell subpopulations and decreased in vitro T-cell proliferation. Additionally, chemotaxis of the neturophils might be reduced.
CVS abnormalities: Ventricular and atrial septal defects, patent foramen ovale, and patent ductus arteriosus may occur in 19% of SDS patients (Myers et al. 2014). Heart failure, possibly secondary to dilated cardiomyopathy, is another common cardiac manifestation in SDS, and it can be severe enough to cause death in the neonatal period (Toiviainen-Salo et al. 2008; Kopel et al. 2011). Histopathology in those cases show myocardial necrosis or fibrosis (Toiviainen-Salo et al. 2008; Burroughs et al. 2009).
Treatment: SDS patients require close monitoring throughout their lifetime for malignancy. Oral pancreatic enzyme supplementation may be necessary, especially early in life. HSCT should be considered in SDS patients with bone marrow failure, myelodysplastic syndrome, and leukemia. Cardiotoxicity, including transient congestive heart failure and long-term cardiac hypokinesia, can occur also following HSCT (Burroughs et al. 2009).
WHIM syndrome
Description: Warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome is an autosomal dominant disorder characterized by difficult-to-treat warts, low IgG levels, recurrent bacterial infections, and neutropenia (Kawai and Malech 2009).
Pathogenesis: WHIM syndrome is caused by mutations in the CXCR4 chemokine receptor gene (OMIM# 193670). This gene is part of a pathway involved in regulation of hematopoiesis, organogenesis, bone marrow homeostasis, and leukocyte motion (Badolato et al. 2012). Mutations in the terminal domain of CXCR4 enhance retention of mature hyper-segmented neutrophils in the bone marrow leading to neutropenia (Kawai and Malech 2009).
Immune abnormalities: The key immunologic features of WHIM syndrome include severe neutropenia. During infections, stress, or granulocyte colony stimulating factor administration, neutrophil numbers in the peripheral blood temporarily increase. WHIM patients might have reduced T- and B-cell numbers as well as hypogammaglobulinemia and abnormal antibody production (Kawai and Malech 2009). Patients with WHIM syndrome are particularly susceptible to papilloma virus infections, leading to warts that can be very extensive and progress to carcinoma (Kawai and Malech 2009).
CVS abnormalities: Few patients with WHIM syndrome have been reported with structural cardiac defects, such as tetralogy of Fallot, possibly because of a role of CXCR4 in cardiac embryogenesis (Badolato et al. 2012).
Treatment: Antibiotic prophylaxis and immunoglobulin replacement can help prevent infections. Papilloma lesions should be aggressively treated and carefully monitored for malignant changes (Kawai and Malech 2009).
MonoMAC syndrome
Description: Several groups recently identified monocytopenia with predisposition to nontuberculosis mycobacterial infections among patients with autosomal dominant mutations in the zinc finger transcription factor GATA2 (OMIM# 137295) (Hsu et al. 2011; Dickinson et al. 2011). Patients are also prone to develop myelodysplastic syndrome and acute myeloid leukemia (Ostergaard et al. 2011).
Pathogenesis: GATA2 is expressed in hematopoietic progenitors, including early erythroid cells, mast cells, megakaryocytes, and also in nonhematopoietic embryonic stem cells, possibly explaining the common hematological abnormalities observed among patients (Hsu et al. 2011).
Immune abnormalities: Patients typically have markedly reduced number of monocytes, B cells, and NK cells in the peripheral blood, resulting in susceptibility to mycobacteria as well as opportunistic fungal infections and disseminated human papillomavirus infections. Abnormal macrophage function in tissue might contribute to the frequent development of pulmonary alveolar proteinosis in these patients. IgG levels and ex vivo T-cell functions are often normal. Erythema nodosum and autoimmunity can be the presenting features (Hsu et al. 2011; Dickinson et al. 2011).
CVS abnormalities: Patients also suffer from primary lymphedema, possibly reflecting the important role of GATA2 in controlling capillary network formation (Hsu et al. 2011, Johnson et al. 2012; Kazenwadel et al. 2012).
Treatment: Patients should be monitored closely for the development of opportunistic infections as well as myelofibrosis and leukemia. HSCT has been curative in some cases.
Chronic granulomatous disease
Description: Chronic granulomatous disease (CGD) is a characterized by inefficient intracellular killing of pathogens resulting in recurrent, severe life threatening infections, inflammation, and granuloma formation (Jones et al. 2008).
Pathogenesis: CGD is caused by mutations in one of 5 genes CYBB (OMIM# 306400), CYBA (OMIM# 608508), NCF1 (OMIM# 608512), NCF2 (OMIM# 608515), and NCF4 (OMIM# 601488) resulting in absent or nonfunctional proteins, gp91 phox, p22 phox, p47 phox, p67 phox, and p40 phox, respectively. Mutations in the CYBB gene cause X-linked CGD and occur in about two-thirds of the patients. All other gene mutations are associated with autosomal recessive CGD. These mutations impair the function of the nicotinamide adenine dinucleotide phosphate-oxidase enzyme complex and prevent production of reactive oxidative species. The impaired oxidative response also enables rapid identification of CGD by flow cytometry analysis, as neutrophils display reduced or absent oxidative burst following ex vivo stimulation (Jones et al. 2008).
Immune abnormalities: Patients are particularly prone to infections by catalase-positive pathogens such as Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia, Aspergillus, Salmonella, and Bacillus Calmette-Guerin. Patients also suffer from persistent inflammation and autoimmunity, particularly involving the gastrointestinal tract, as well as formation of multiple granulomas, which may even cause obstruction of viscera such as the stomach, urinary track, etc. (Marciano et al. 2004; Jones et al. 2008).
CVS abnormalities: A large retrospective chart review of CVS disease among 229 patients with CGD followed at the U.S. National Institute of Health, identified 2 patients with coronary artery disease and another patient with aortic and mitral valve regurgitations (Leiding et al. 2013). Interestingly, the 3 patients with CVS abnormalities had p47 PHOX mutations. One of these patients was also found to have cerebral aneurysms, diagnosed during family screening. Affected patients had other risk factors for CVS abnormalities such as diabetes and dysregulated cholesterol; hence, the direct contribution of defective reactive oxidative species production in CVS disease development needs to be further studied. Interestingly, a recent study identified increased nitric oxide, a potent vasodilator molecule produced by endothelial cells, and significantly higher flow-mediated dilatation among 30 patients with CGD (Loffredo et al. 2013). The clinical significance of these findings for the development of CVS abnormalities is still not clear.
Treatment: Patients with CGD should receive antibacterial and antifungal prophylaxis. Infections and granulomas should be treated promptly with antibiotics and immunosuppressive medications, respectively. HSCT, from family related or unrelated HLA-matched donors, can cure CGD. Currently, human trials of gene therapy for CGD are underway.
Well-defined syndromes with immunodeficiency
DiGeorge syndrome
Description: DGS, estimated to occur in 1:3000 live births, is characterized by variable combinations of neonatal hypocalcaemia, thymus hypoplasia, and conotruncal defects that disrupt the cardiac outflow tract (OMIM# 188400). De novo or autosomal dominant inheritance deletion of chromosome 22q11.2 are found among 80%–90% of the patients, although haplo-insufficiency of others chromosomes such as 10p13 or 4q21.3-q25 have also been implicated. Other features commonly identified among patients with 22q11.2 microdeletion include renal anomalies, ophthalmologic abnormalities, developmental delay, learning disabilities, schizophrenia, and bipolar disorders as well skeletal defects and short stature. Palate abnormalities are also common, resulting in velocardiofacial syndrome (OMIM#192430). Patients also have typical dysmorphic facial features including micrognathia, low set ears, telecanthus with short palpebral fissures, upward or downward slanting eyes with short philtrum, and a relatively small mouth (Kobrynski and Sullivan 2007).
Pathogenesis: DGS is thought to be secondary to disrupted cervical neural crest migration into the derivatives of the 3rd and 4th pharyngeal arches and pouches. Among patients with 22q11.2 deletion, haplo-insufficiency of the TBX1 gene, which encodes a transcription factor important for neural crest cells migration, is thought to cause the majority of the features (Kobrynski and Sullivan 2007).
Immune abnormalities: DGS is the most common human T-cell immune deficiency. The abnormalities of the thymus impair thymocytes development and selection, often manifesting with diminished T-cell counts in the peripheral blood, although the responses of T cells to stimulation is often preserved. Indeed in the majority of patients with DGS, the laboratory T-cell abnormalities are not associated with increased susceptibility to infections. Moreover, the number of T cells increases during infancy and childhood among most patients. “Complete DGS” with thymus aplasia and practically absent T cells has been reported in <1% of the patients. However, because of the potential severity and lethality of this condition, as well as the lack of correlation with nonimmune abnormalities, complete DGS should be excluded in all patients. Severe thymus defects cause marked reduction of new T-cell production, resulting in low TREC. Importantly, TREC, which can be measured in peripheral blood using PCR, enable detection of severe DGS in newborn screening programs recently implemented among many counties. Severe DGS can also manifest as Omenn syndrome with misleading normal or even elevated T-cell numbers. Few patients have also been identified with hemophagocytic lymphohistiocytosis. Autoimmune conditions, resulting in hematological cytopenias or rheumatologic complications, are also common in patients with DGS. Infrequently, patients with DGS also display humoral immunodeficiency with reduced production of IgG or specific antibodies (Kobrynski and Sullivan 2007; Gennery 2012).
CVS abnormalities: Approximately 80% of patients with DGS and 22q11.2 deletion syndrome have various congenital heart defects (Momma 2010). The heart disease typically manifests as cardiac outflow tract anomalies related to impaired embryogenesis. The most common defects are tetralogy of Fallot, ventricular septal defect, interrupted aortic arch, and truncus arteriosus. Other reported anomalies include right aortic arch and aberrant right subclavian artery, atrial septum defect, pulmonary valve stenosis, double outlet right ventricles, coarctation of the aorta, great arteries transposition, etc. (Momma 2010).
Treatment: Management of DGS depends on the clinical phenotype. Cardiac defects may require early surgical intervention, whereas the hypocalcaemia often can be treated with calcium supplementation (Kobrynski and Sullivan 2007). Antimicrobial prophylaxis and immunoglobulin replacement should be considered for the immunodeficiency (Gennery 2012) as well as adherence to cytomegalovirus-negative irradiated blood products. Yet for most patients with DGS, the normal or near-normal immune system does enable administration of live viral vaccines (Al-Sukaiti et al. 2010). Allogeneic thymus tissue transplantation or HSCT have been used for the rare “complete DGS” with variable success.
CHARGE syndrome
Description: A multiple congenital cardiac and immune anomaly disorder (OMIM# 214800) characterized by coloboma of the eye, heart anomalies, atresia choanae, retardation of mental and physical development, genital anomalies, and ear abnormalities/deafness is known as CHARGE. There is much phenotypic variability among patients with CHARGE syndrome, and the clinical diagnostic criteria have been revised several times (Bergman et al. 2011).
Pathogenesis: CHARGE syndrome typically arises from autosomal dominant mutations in transcription regulator chromodomain helicase DNA-binding protein-7 gene. During embryonic development, the product of this gene is thought to be essential for normal formation of multipotent migratory neural crest cells. Further, its expression in the central nervous system, semicircular canals, and neural crest of the pharyngeal arches explains some of the diverse tissue abnormalities found in CHARGE syndrome (Gennery et al. 2008; Bergman et al. 2011).
Immune abnormalities: Mild to moderate T-cell deficiencies, including Omenn syndrome, are infrequently observed among patients with CHARGE syndrome. Isolated antibody deficiency is also rarely found in CHARGE syndrome (Writzl et al. 2007; Gennery et al. 2008; Jyonouchi et al. 2009).
CVS abnormalities: Congenital heart lesions occur in 75%–80% of patients (Blake and Prasad 2006). Conotruncal heart defects, especially tetralogy of Fallot, are the most common manifestation, similar to DGS. However, many lesions have been reported including atrioventricular canal defects, aortic arch defects, and patent ductus arteriosus, as well as atrial and ventricular septal defects (Blake and Prasad 2006).
Treatment: Surgical intervention may be required for choanal atresia and cardiac defects. All CHARGE syndrome patients should have ophthalmological follow-up as well as hearing screens and hearing aids if necessary. Potential immunodeficiency should be screened for and managed accordingly (Blake and Prasad, 2006).
Wiskott–Aldrich syndrome
Description: Wiskott–Aldrich syndrome (WAS) is an X-linked disorder characterized by thrombocytopenia, severe eczema, and recurrent infections (OMIM# 301000). Thrombocytopenia may manifest early as petechiae, bruising, bloody diarrhea, or excessive bleeding after circumcision. The platelets are typically small in size (Ochs et al. 2009).
Pathogenesis: WAS is caused by mutations in the WAS gene located on the X chromosome. Accordingly, it is predominantly found in males, although rarely females with nonrandom X inactivation may develop a similar phenotype. The WAS gene codes for the WAS protein, a cytoskeletal regulator expressed in hematopoietic cells (Ochs et al. 2009; Bouma et al. 2009). WAS protein is involved in cell signaling, migration, and immune synapse formation (Ochs et al. 2009; Bouma et al. 2009). Many different types of mutations within this gene cause WAS; however, clear correlation between the mutations and phenotypic severity have not been established.
Immune abnormalities: Infections, including recurrent otitis media, bacterial pneumonias, and skin infections are common among patients suffering from WAS (Ochs et al. 2009). Opportunistic infections such as Pneumocystis jiroveci pneumonia can also occur, reflecting the combined T- and B-cell dysfunction in this condition. There is often decreased numbers of circulating T cells, particularly CD8+ T cells, as well as diminished T-cell function. IgG and IgM may be normal or decreased, whereas IgA and IgE are typically elevated (Ochs et al. 2009). Infrequently, elevated IgM and reduced IgG, reminiscent of hyper-IgM syndrome can be found as well as abnormal responses to polysaccharide antigens (Ochs et al. 2009). Impaired immune cell migration and defects in the innate immune system are also observed in WAS (Bouma et al. 2009). Other manifestations of this syndrome include autoimmune disorders, such as hemolytic anemia or immune-mediated thrombocytopenia, which further complicates the diagnosis and management of the platelet disorder (Ochs et al. 2009). Increased susceptibility to malignancies, including lymphoma and leukemia, as well as solid tumors is noted.
CVS abnormalities: Aneurysm formation as well as small, medium, and large (including aorta) vessel vasculitis are found in few patients with WAS. These include a 24-year-old patient who developed necrotizing vasculitis of small and medium sized arteries, resulting in aneurysms of his renal, hepatic, and superior mesenteric arteries (McCluggage et al. 1999) and a 7-year-old boy with aortic root dilatation (Ono et al. 2009). Pathology in the latter child showed aortitis with inflammatory infiltration and necrotic areas. Additional WAS patients with aortic aneurysms have been described (Pellier et al. 2011). The pathogenesis for the vasculitis is not clear, yet it might precede the thrombocytopenia (Mahlaoui et al. 2013).
Treatment: Patients with WAS typically require antimicrobial prophylaxis to prevent infections and platelet transfusions to treat significant bleeding episodes (Ochs et al. 2009). Immunosuppressive therapy may be necessary to treat autoimmune manifestations particularly the thrombocytopenia (Ochs et al. 2009). Splenectomy was previously used for severe thrombocytopenia; however, such procedure increases the risk for infections and it does not prevent malignancy. HSCT, from related or unrelated HLA-matched donors has been shown to be highly beneficial for patients suffering from WAS, particularly if performed at an early age. Currently, human trials of gene therapy for WAS are underway.
Roifman syndrome
Description: Roifman syndrome (OMIM# 300258) is characterized by antibody deficiency, spondyloepiphyseal dysplasia, growth retardation, and retinal dystrophy (Roifman 1999). Associated manifestations include intellectual disability, dysmorphic features, and hypogonadism.
Pathogenesis: The gene responsible for Roifman Syndrome has not yet been identified.
Immune abnormalities: Roifman Syndrome is associated with humoral immunodeficiency, particularly poor specific antibody response. This results in recurrent sinopulmonary infections including otitis media and pneumonia (Roifman 1999).
CVS abnormalities: A 14-year-old boy with Roifman Syndrome was reported to suffer from left ventricular noncompaction and heart failure (Mandel et al. 2001).
Treatment: Patients are managed with immunoglobulin replacement and close monitoring.
Schimke immuno-osseus dysplasia
Description: Schimke immuno-osseus dysplasia (SIOD) is an autosomal recessive disorder characterized by spondyloepiphysial dysplasia, immunodeficiency, facial dysmorphism, and progressive renal disease (OMIM# 242900). Other features include skin lesions, hypothyroidism, headaches, cerebral ischemia, and enteropathy (Boerkoel et al. 2000). There appears to be 2 forms of SIOD: juvenile onset and the more severe infantile onset (Boerkoel et al. 2000).
Pathogenesis: SIOD is caused by biallelic loss-of-function mutations in the SMARCAL1 gene (Clewing et al. 2007). This gene encodes a protein involved in chromatin remodeling, but the pathogenic mechanism of SIOD is still unclear (Clewing et al. 2007).
Immune abnormalities: Immune dysfunction in SIOD is primarily T-cell deficiency and dysfunction resulting in increased susceptibility to opportunistic fungal, viral, and bacterial infections (Boerkoel et al. 2000; Lev et al. 2009).
CVS abnormalities: Vascular abnormalities occur in many patients, including transient ischemic attacks and (or) cerebral ischemia, putatively secondary to early onset, and progressive arteriosclerosis (Boerkoel et al. 2000; Clewing et al. 2007). A recent study among more than 50% of SIOD patients found arteriosclerosis associated with vascular intimal and medial hyperplasia, smooth muscle cell hyperplasia, and fragmented and disorganized elastin fibers (Morimoto et al. 2012). Few patients were reported to have cerebral artery stenosis and moya-moya phenomenon (Boerkoel et al. 1998).
Treatment: Dialysis and renal transplant are used for the progressive renal failure (Boerkoel et al. 2000).
Hyper-IgE syndromes
Description: Hyper-IgE Syndrome (HIES) represents a group of disorders characterized by recurrent skin and lung infections, eczema, and elevated IgE levels (Heimall et al. 2010). An autosomal dominant form (OMIM# 147060), also known as Job's Syndrome, is associated with distinctive coarse facial features, delayed shedding of primary teeth, frequent bone fractures, spinal deformities, vascular anomalies, and development of lymphoma (Heimall et al. 2010). Pulmonary infections often result in pneumatoceles and (or) bronchiectasis. The autosomal recessive form (OMIM# 243700) is often associated with a more profound immunodeficiency resulting in susceptibility to viral infections, particularly papilloma and herpes viruses, autoimmunity, sclerosing cholangitis, and vasculitis (Heimall et al. 2010; Alsum et al. 2013).
Pathogenesis: Autosomal dominant HIES can be caused by mutations in the STAT3 gene (Heimall et al. 2010). The STAT3 protein is part of the JAK–STAT signal-transducing pathway, responsible for activating gene transcription and translation (Heimall et al. 2010). Autosomal recessive HIES is caused primarily by mutations in DOCK8, although in a few patients defects in TYK2 were identified. Somatic mutations in STAT3 have also been recently identified among patients with a milder phenotype (Hsu et al. 2013).
Immune abnormalities: Markedly elevated IgE levels are common among patients with HIES; however, similar levels might be found among patients with atopic conditions as well as other immune deficiencies. Patients with HIES might also have fluctuating as well as normal or near normal IgE levels, making the IgE level an unreliable diagnostic criteria. Some patients might have impaired antibody responses or decreased IgG levels. The autoimmunity in HIES is attributed to decreased IL-17 producing T-helper cells. Importantly, severe T- and B-cell defects can occur early in life in some patients with DOCK8 mutations.
CVS abnormalities: Vascular abnormalities involving arteries or veins of the brain, lungs, heart, and skin as well as coronary and aortic aneurysms have been identified among patients with STAT3 defects (Yavuz and Chee, 2010). Similar vascular abnormalities including giant aortic aneurysm, brain infarction, and moya-moya syndrome were identified in patients with DOCK8 mutations (Alsum et al. 2013). It is still not clear whether the vascular lesions are secondary to congenital angiogenesis defects post-infectious inflammation or to autoimmunity.
Treatment: Patients with abnormal antibody production should receive immunoglobulin replacement. Patients with severe autosomal recessive HIES can benefit from HSCT.
Immunodeficiency with centromeric instability and facial anomalies
Description: Immunodeficiency with centromeric instability and facial anomalies (ICF) syndrome (OMIM# 242860) is an autosomal recessive disease. ICF can be caused by mutations in the DNA methyltransferase 3B gene at chromosome 20q11.2 (designated ICF1) or the zinc-finger and BTB domain-containing 24 gene (ICF2). Patients have mild facial anomalies including a round face, flat nasal bridge, hypertelorism, epicanthus, up-turned nose, macroglossia, micrognathia, and low-set ears. The majority of ICF patients have delayed walking and speech, and some may suffer from intellectual disabilities. Diagnosis of ICF can be established by demonstrating excess deletions and peri-centromeric breaks of chromosomes 1, 9, and 16 in metaphases of stimulated peripheral blood lymphocytes (Weemaes et al. 2013).
Pathogenesis: Hypomethylation of various genes, including those important for immune and neurological development, as well as satellite DNA located in the pericentromeric regions of chromosomes 1, 9, and 16 may lead to the diverse clinical and laboratory features (Weemaes et al. 2013).
Immune abnormalities: Severe infections including pneumonia, sepsis, as well as opportunistic infections caused by pathogens such as Candida albicans, Pneumocystis jiroveci, and JC virus may occur in ICF patients. Hypo- or agammaglobulinemia is found among most patients with ICF. Patients’ circulating B cells contain an increased proportion of immature cells and reduced memory cells. Additionally, B cells are more prone to undergo apoptosis upon in vitro activation. The numbers of CD3+, CD4+, and CD8+ cells are usually normal. However, with age, some patients exhibit T-cell lymphopenia and reduced proliferation of T cells following ex vivo stimulation. Few patients also develop neutropenia in the second decade of life (Weemaes et al. 2013).
CVS abnormalities: Ventricular and atrial septum defects were reported in few patients.
Treatment: Antibody deficiency should be treated with immunoglobulin replacement. HSCT was performed in few patients because of severe infections or myelodysplasia with some success (Weemaes et al. 2013).
Hepatic veno-occlusive disease with immunodeficiency
Description: Hepatic veno-occlusive disease with immunodeficiency (VODI) syndrome (OMOM# 235550) was first described in Lebanese descendants in Australia, followed by descriptions from other continents and populations. VODI is an autosomal recessive disease caused by mutations in the SP110 gene (Roscioli et al. 2006). Some patients suffer from microcephaly, leukodystrophy, multiple small cerebral calcifications, and neuro-developmental defects, as well as pulmonary fibrosis, hepatosplenomegaly, and thrombocytopenia. Histological and ultrasound-Doppler can establish the diagnosis of hepatic sinusoidal obstructive syndrome.
Pathogenesis: The molecular pathogenesis of VODI is not clear; however, it might be related to abnormalities in the PML nuclear body protein, a transcriptional factor that enhances transcription of genes with retinoic acid response elements (Roscioli et al. 2006).
Immune abnormalities: Multiple infections with pathogens such as Pneumocystis jiroveci, cytomegalovirus, and Candida often occur in infancy, reflecting a combined T- and B-cell immunodeficiency. Severe hypogammaglobulinemia as well as reduced IgM and IgA are also commonly found. Histological evaluations often reveal absent lymph nodes’ germinal centers and tissue plasma cells. Neutropenia can also occur (Roscioli et al. 2006).
CVS abnormalities: In addition to the characteristic hepatic veno-occlusive disease, left atrial endocardial fibrosis can occur (Roscioli et al. 2006).
Treatment: Patients should receive immunoglobulin replacement and Pneumocystis jiroveci prophylaxis. HSCT was successful among few patients (Ganaiem et al. 2013).
Facial dysmorphism, immunodeficiency, livedo, and short stature (FILS syndrome)
Description: Recently, a single large consanguineous French kindred was identified with mild facial dysmorphism, immunodeficiency, livedo, and short stature caused by mutations in the DNA polymerase ɛ gene (POLE) (Pachlopnik Schmid et al. 2012). Few patients also display bone dysplasia with lacunar lesions, cortical thickening, and modeling defects of long bone diaphysis as well as metaphyseal striae. Growth impairment can be observed during early childhood impairing adult height.
Pathogenesis: The precise mechanism is still not clear; however, it is hypothesized that absence of DNA polymerase ɛ impedes proliferation and G1- to S-phase progression in lymphocytes, chondrocytes, and osteoblasts (Pachlopnik Schmid et al. 2012).
Immune abnormalities: The immunodeficiency often manifests early in life with recurrent respiratory tract infections resulting in bronchiectasis. Some patients also suffer recurrent meningitis caused by Streptococcus pneumonia. Immune evaluation of the patients shows decreased IgM and IgG2 levels, reduced isohemagglutinin titers, and a predominant lack of antibodies to polysaccharide antigens. Patients may have low memory B- and naive T-cell counts as well as decreased T-cell proliferation (Pachlopnik Schmid et al. 2012).
CVS abnormalities: Livedo on the cheeks, forearms, and (or) legs is present in most patients and can sometimes be noticed from birth. With increasing age, telangiectasia can be observed on the cheeks (Pachlopnik Schmid et al. 2012).
Treatment: Patients might benefit from prophylactic antibiotics. Whether immunoglobulin replacement is beneficial for these patients is not clear.
Disease of immune dysregulation
X-linked lymphoproliferative disease
Description: X-linked lymphoproliferative disease (XLP) is characterized by severe immune dysregulation, often triggered by EBV infection (OMIM# 308240). Some patients suffer recurrent infections, whereas others might present with fulminant infectious mononucleosis and liver failure as well as EBV or non-EBV induced hemophagocytic lymphohistiocytosis and lymphoma (Booth et al. 2011). The phenotype is highly variable, even within the same family. Other less common presenting features include aplastic anemia, vasculitis, colitis, and chronic gastritis (Booth et al. 2011).
Pathogenesis: XLP disease is caused by mutations in the SH2D1A gene, located on the X chromosome; hence, the disease affects males. The gene encodes SAP, a protein that helps regulate immune function in T, NK, and possibly B cells (Booth et al. 2011).
Immune abnormalities: In XLP, the numbers of circulating T cells are normal, but their function is impaired. Some patients display hypogammaglobulinemia or dysgammaglobulinemia. Low numbers of NK cells are also common (Booth et al. 2011).
CVS abnormalities: Systemic vasculitis has been reported in few patients with XLP. These include small and medium vessel multi-organ lymphocytic vasculitis (Dutz et al. 2001).
Treatment: Antibody deficiency should be treated with immunoglobulin replacement. Because of the risk for hemophagocytic lymphohistiocytosis and lymphoma, preventive HSCT with appropriate donors should be considered, particularly in young patients (Booth et al. 2011).
FADD deficiency
Description: FADD deficiency (OMIM# 613759) can cause autoimmune lymphoproliferative syndrome (ALPS) (Bolze et al. 2010). Patients suffer recurrent bacterial and viral infections, hepatic dysfunction, functional asplenia, encephalopathy, and cardiac malformations.
Pathogenesis: FADD deficiency is an autosomal recessive disease caused by mutations in the FADD gene on chromosome 11q13.3 (Bolze et al. 2010). The FADD gene encodes an adaptor molecule that interacts with FAS, an apoptosis-inducing receptor molecule. The FADD protein is also involved in autophagy, inflammation, innate immunity, cell proliferation, and tumor development. Mutations in FADD lead to impaired FAS-dependent apoptosis and immune dysregulation (Bolze et al. 2010).
Immune abnormalities: FADD deficiency is associated with elevated double negative T cells, and elevated IL-10 and Fas-Ligand levels. FADD-deficient patients display Howell–Jolly bodies in their blood smears, indicating functional asplenia, which may explain the susceptibility of patients to severe and invasive bacterial infections (Bolze et al. 2010).
CVS abnormalities: Two of the 4 patients with FADD deficiency had cardiac malformations. One had pulmonary atresia with a ventricular septal defect and the other had a left-sided superior vena cava, draining into the left atrium (Bolze et al. 2010). Mice are known to express FADD in the myocardium and mice with FADD deficiency have embryonic defects, suggesting that FADD deficiency might affect cardiogenesis (Bolze et al. 2010).
Treatment: At present there is no curative therapy for this syndrome. Only one of the 4 described patients is still alive (Bolze et al. 2010).
Aicardi–Goutières syndrome
Description: Aicardi–Goutières syndrome (AGS) is characterized by familial encephalopathy resembling intrauterine viral infection (OMIM# 225750). Patients suffer from varying severities of psychomotor retardation, microcephaly, dystonic posturing, and spasticity. Chilblain-like skin lesions and congenital glaucoma may also be present. Brain imaging often reveals intra-cerebral calcifications, particularly in the basal ganglia, cerebral atrophy, and leukodystrophy. The syndrome manifests in infancy, and death often occurs early in childhood; however, milder forms and survival into adulthood can occur. AGS is caused by mutations in different genes including TREX1, RNASEH2A, RNASEH2B and RNASEH2C, as well as ADAR1 and SAMHD1 (Kavanagh et al. 2008).
Pathogenesis: AGS is thought to be caused by impaired clearance of nucleic acids and abnormal intracellular accumulation of single-stranded DNA species, which trigger an inappropriate type I interferon-mediated immune response leading to vasculitis and vascular changes (Kavanagh et al. 2008).
Immune abnormalities: Patients may suffer from various autoimmune manifestations including arthritis, thrombocytopenia, and elevated transaminases, and they may also develop autoantibodies. Typically lymphocytosis and elevated interferon-α can be found in the cerebrospinal fluid, mimicking congenital transplacentally acquired infection, which needs to be excluded. Pterins in cerebrospinal fluid can help establish the diagnosis (Kavanagh et al. 2008).
CVS abnormalities: Defects in TREX1 are associated with transient ischemic attacks and strokes (Kavanagh et al. 2008). Histological assessments show obliterative vasculopathy in the brain, whereas fluorescein angiograms demonstrate retinal vasculopathy. Additionally, small vessel gastrointestinal bleeding and renal arteriolonephrosclerosis have been observed. Few patients with SAMHD1 deficiency have stenosis of carotid and intracranial vessels leading to stroke (Kavanagh et al. 2008).
Treatment: Recognizing AGS as an autoimmune/autoinflammatory condition has led to the use of various immune modulating agents, although it is difficult to assess their efficacy. Alternative strategies exploring blocking antibodies to interferon-α, reverse transcriptase inhibition, or B-cell depletion are currently being developed (Crow et al. 2014).
Autoinflammatory disorders
HOIL1 deficiency
Description: Mutations in HOIL1 (RBCK1) can cause an autosomal recessive disease characterized by persistent infections, auto-inflammation, severe failure to thrive, liver fibrosis with hepatocytes vacuoles, and amylopectin-like deposits (Boisson et al. 2012).
Pathogenesis: Three patients from 2 unrelated families were shown to carry loss-of-function mutations in HOIL1, a component of the linear ubiquitination chain assembly complex. The defect impairs fibroblasts NF-κB activation in response to IL-1β and TNF-α. In contrast, the patients’ mononuclear leukocytes, particularly monocytes, were hyper-responsive to IL-1β.
Immune abnormalities: Patients are susceptible to pyogenic bacterial and CMV infections. Auto-inflammation may manifest in infancy with recurrent fevers, severe eczematous rash, lymphadenopathy, hepato-splenomegaly, inflammatory bowel disease, and elevated inflammatory cytokines. Laboratory evaluations demonstrate increased IgA, decreased memory B cells and minor defects in antibody production following vaccinations (Boisson et al. 2012).
CVS abnormalities: Patients display muscular amylopectinosis, evident already in infancy, which contributes to progressive muscular atrophy as well as severe dilated cardiomyopathy and congestive cardiac failure. Amylopectin-like deposits are found in cardiomyocytes as well as gut muscular mucosa and skeletal muscle cells (Boisson et al. 2012).
Treatment: Supportive therapy for infections and immune dysregulation is necessary. One patient, who died 3 years after successful HSCT, had sub-endocardial fibrosis with cytoplasmic vacuolization of myocardiocytes and modest mononuclear cell infiltrates, indicative of chronic myocarditis (Boisson et al. 2012).
Conclusion
In conclusion, as described in this comprehensive review, the expanding spectrum of PID requires increased alertness to the possibility of CVS involvement as an important contributor to the diagnosis and management of these patients.
REFERENCES
Abdollahpour H., Appaswamy G., Kotlarz D., Diestelhorst J., Beier R., Schaffer A.A., Gertz E.M., Schambach A., Kreipe H.H., Pfeifer D., Engelhardt K.R., Rezaei N., Grimbacher B., Lohrmann S., Sherkat R., and Klein C. The phenotype of human STK4 deficiency Blood. 2012 119 15 3450 -3457
Albert M.H., Gennery A.R., Greil J., Cale C.M., Kalwak K., Kondratenko I., Mlynarski W., Notheis G., Führer M., Schmid I., and Belohradsky B.H. Successful SCT for Nijmegen breakage syndrome Bone. Marrow. Transpl. 2010 45 4 622 -626
Aleman K., Noordzij J.G., De Groot R., Van Dongen J.J., and Hartwig N.G. Reviewing Omenn syndrome Eur. J. Pediatr. 2001 160 12 718 -725
Al-Sukaiti N., Reid B., Lavi S., Al-Zaharani D., Atkinson A., Roifman C.M., and Grunebaum E. Safety and efficacy of measles, mumps, and rubella vaccine in patients with DiGeorge syndrome J. Allergy. Clin. Immunol. 2010 126 4 868 -869
Alsum Z., Hawwari A., Alsmadi O., Al-Hissi S., Borrero E., Abu-Staiteh A., Khalak H.G., Wakil S., Eldali A.M., Arnaout R., Al-Ghonaium A., Al-Muhsen S., Al-Dhekri H., Al-Saud B., and Al-Mousa H. Clinical, immunological and molecular characterization of DOCK8 and DOCK8-like deficient patients: single center experience of twenty-five patients J. Clin. Immunol. 2013 33 1 55 -67
Badolato R., Dotta L., Tassone L., Amendola G., Porta F., Locatelli F., Notarangelo L.D., Bertrand Y., Bachelerie F., and Donadieu J. Tetralogy of fallot is an uncommon manifestation of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome J. Pediatr. 2012 161 4 763 -765
Banka S., Chervinsky E., Newman W.G., Crow Y.J., Yeganeh S., Yacobovich J., Donnai D., and Shalev S. Further delineation of the phenotype of severe congenital neutropenia type 4 due to mutations in G6PC3 Eur. J. Hum. Genet. 2011 19 1 18 -22
Bastianon V., Giglioni E., Businco L., Fiorilli M., and Chessa L. Cardiac anomalies in ataxia-telangiectasia Am. J. Dis. Child. 1993 147 1 20 -21
Begin P., Patey N., Mueller P., Rasquin A., Sirard A., Klein C., Haddad É., Drouin É., and Le Deist F. Inflammatory bowel disease and T cell lymphopenia in G6PC3 deficiency J. Clin. Immunol. 2013 33 3 520 -525
Bergman J.E.H., Janssen N., Hoefsloot L.H., Jongmans M.C.J., Hofstra R.M.W., and Van Ravenswaaij-Arts C.M.A. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype J. Med. Genet. 2011 48 5 334 -342
Blake K.D. and Prasad C. CHARGE syndrome Orphanet. J. Rare. Dis. 2006 1 34
Boerkoel C.F., Nowaczyk M.J.M., Blaser S.I., Meschino W.S., and Weksberg R. Schimke immunoosseous dysplasia complicated by moyamoya phenomenon Am. J. Med. Genet. 1998 78 2 118 -122
Boerkoel C.F., O'neill S., Andr J.L., Benke P.J., Bogdanov R., Bulla M., Burguet A., Cockfield S., Cordeiro I., Ehrich J.H.H., FrNd S., Geary D.F., Ieshima A., Illies F., Joseph M.W., Kaitila I., Lama G., Leheup B., Ludman M.D., Mcleod D.R., Medeira A., Milford D.V., Rm T., Rener-Primec Z., Santava A., Santos H.G., Schmidt B., Smith G.C., Spranger J., Zupancic N., and Weksberg R. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature Eur. J. Pediatr. 2000 159 1–2 1 -7
Boisson B., Laplantine E., Prando C., Giliani S., Israelsson E., Xu Z., Abhyankar A., Israël L., Trevejo-Nunez G., Bogunovic D., Cepika A.-M., Macduff D., Chrabieh M., Hubeau M., Bajolle F., Debré M., Mazzolari E., Vairo D., Agou F., Virgin H.W., Bossuyt X., Rambaud C., Facchetti F., Bonnet D., Quartier P., Fournet J.-C., Pascual V., Chaussabel D., Notarangelo L.D., Puel A., Israël A., Casanova J.-L., and Picard C. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency Nat. Immunol. 2012 13 12 1178 -1186
Bolze A., Byun M., Mcdonald D., Morgan N.V., Abhyankar A., Premkumar L., Puel A., Bacon C.M., Rieux-Laucat F., Pang K., Britland A., Abel L., Cant A., Maher E.R., Riedl S.J., Hambleton S., and Casanova J.-L. Whole-exome-sequencing-based discovery of human FADD deficiency Am. J. Hum. Genet. 2010 87 6 873 -881
Booth C., Gilmour K.C., Veys P., Gennery A.R., Slatter M.A., Chapel H., Heath P.T., Steward C.G., Smith O., O'meara A., Kerrigan H., Mahlaoui N., Cavazzana-Calvo M., Fischer A., Moshous D., Blanche S., Pachlopnik Schmid J., Latour S., De Saint-Basile G., Albert M., Notheis G., Rieber N., Strahm B., Ritterbusch H., Lankester A., Hartwig N.G., Meyts I., Plebani A., Soresina A., Finocchi A., Pignata C., Cirillo E., Bonanomi S., Peters C., Kalwak K., Pasic S., Sedlacek P., Jazbec J., Kanegane H., Nichols K.E., Hanson I.C., Kapoor N., Haddad E., Cowan M., Choo S., Smart J., Arkwright P.D., and Gaspar H. B. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease Blood. 2011 117 1 53 -62
Bouma G., Burns S.O., and Thrasher A.J. Wiskott-Aldrich Syndrome: immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation Immunobiology. 2009 214 9–10 778 -790
Bousfiha A.A., Jeddane L., Ailal F., Al Herz W., Conley M.E., Cunningham-Rundles C., Etzioni A., Fischer A., Franco J.L., Geha R.S., Hammarström L., Nonoyama S., Ochs H. D., Roifman C.M., Seger R., Tang M.L.K., Puck J.M., Chapel H., Notarangelo L.D., and Casanova J.-L. A phenotypic approach for IUIS PID classification and diagnosis guidelines for clinicians at the bedside J. Clin. Immunol. 2013 33 6 1078 -1087
Boztug K., Appaswamy G., Ashikov A., Schäffer A.A., Salzer U., Diestelhorst J., Germeshausen M., Brandes G., Lee-Gossler J., Noyan F., Gatzke A.-K., Minkov M., Greil J., Kratz C., Petropoulou T., Pellier I., Bellanné-Chantelot C., Rezaei N., Mönkemöller K., Irani-Hakimeh N., Bakker H., Gerardy-Schahn R., Zeidler C., Grimbacher B., Welte K., and Klein C. A syndrome with congenital neutropenia and mutations in G6PC3 N. Engl. J. Med. 2009 360 1 32 -43
Bruckmann C., Lindner W., Roos R., Permanetter W., Haas R.J., Haworth S.G., and Belohradsky B.H. Severe pulmonary vascular occlusive disease following bone marrow transplantation in Omenn syndrome Eur. J. Pediatr. 1991 150 4 242 -245
Burroughs L., Woolfrey A., and Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol. Clin. North Am. 2009 23 2 233 -248
Cavadini P., Vermi W., Facchetti F., Fontana S., Nagafuchi S., Mazzolari E., Sediva A., Marrella V., Villa A., Fischer A., Notarangelo L.D., and Badolato R. AIRE deficiency in thymus of 2 patients with Omenn syndrome J. Clin. Invest. 2005 115 3 728 -732
Chrzanowska K.H., Gregorek H., Dembowska-Baginska B., Kalina M.A., and Digweed M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare. Dis. 2012 7 13
Clewing J.M., Antalfy B.C., Lucke T., Najafian B., Marwedel K.M., Hori A., Powel R. M., Safo Do A.F., Najera L., Santacruz K., John Hicks M., Armstrong D.L., and Boerkoel C.F. Schimke immuno-osseous dysplasia: a clinicopathological correlation J. Med. Genet. 2007 44 2 122 -130
Conley M.E., Dobbs A.K., Farmer D.M., Kilic S., Paris K., Grigoriadou S., Coustan-Smith E., Howard V., and Campana D. Primary B cell immunodeficiencies: comparisons and contrasts Annu. Rev. Immunol. 2009 27 199 -227
Crow Y.J., Vanderver A., Orcesi S., Kuijpers T.W., and Rice G.I. Therapies in Aicardi-Goutieres syndrome Clin. Exp. Immunol. 2014 175 1 1 -8
Cunningham-Rundles C. The many faces of common variable immunodeficiency Hematol. Am. Soc. Hematol. Educ. Prog. 2012 2012 301 -305
Dickinson R.E., Griffin H., Bigley V., Reynard L.N., Hussain R., Haniffa M., Lakey J. H., Rahman T., Wang X.-N., Mcgovern N., Pagan S., Cookson S., Mcdonald D., Chua I., Wallis J., Cant A., Wright M., Keavney B., Chinnery P.F., Loughlin J., Hambleton S., Santibanez-Koref M., and Collin M. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency Blood. 2011 118 10 2656 -2658
Dror Y., Ginzberg H., Dalal I., Cherepanov V., Downey G., Durie P., Roifman C.M., and Freedman M.H. Immune function in patients with Shwachman-Diamond syndrome Br. J. Haematol. 2001 114 3 712 -717
Dror Y., Donadieu J., Koglmeier J., Dodge J., Toiviainen-Salo S., Makitie O., Kerr E., Zeidler C., Shimamura A., Shah N., Cipolli M., Kuijpers T., Durie P., Rommens J., Siderius L., and Liu J.M. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome Ann. NY Acad. Sci. 2011 1242 40 -55
Dutz J.P., Benoit L., Wang X., Demetrick D.J., Junker A., de Sa D., and Tan R. Lymphocytic vasculitis in X-linked lymphoproliferative disease Blood. 2001 97 1 95 -100
Ege M., Ma Y., Manfras B, Kalwak K, Lu H., Lieber M.R., Schwarz K., and Pannicke U. Omenn syndrome due to ARTEMIS mutations Blood. 2005 105 11 4179 -4186
Feske S., Picard C., and Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1 Clin. Immunol. 2010 135 2 169 -182
Finsterer J. and Frank M. Haematological features in Barth syndrome Curr. Opin. Hematol. 2013 20 1 36 -40
Ganaiem H., Eisenstein E.M., Tenenbaum A., Somech R., Simanovsky N., Roscioli T., Weintraub M., and Stepensky P. The role of hematopoietic stem cell transplantation in SP110 associated veno-occlusive disease with immunodeficiency syndrome Pediatr. Allergy. Immunol. 2013 24 3 250 -256
Gennery A.R. Immunological aspects of 22q11.2 deletion syndrome Cell. Mol. Life. Sci. 2012 69 1 17 -27
Gennery A.R., Slatter M.A., Rice J., Hoefsloot L.H., Barge D., Mclean-Tooke A., Montgomery T., Goodship J.A., Burt A.D., Flood T.J., Abinun M., Cant A.J., and Johnson D. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome Clin. Exp. Immunol. 2008 153 1 75 -80
Grouhi M., Dalal I., Nisbet-Brown E., and Roifman C.M. Cerebral vasculitis associated with chronic mucocutaneous candidiasis J. Pediatr. 1998 133 4 571 -574
Grunebaum E., Bates A., and Roifman C.M. Omenn syndrome is associated with mutations in DNA ligase IV J. Allergy. Clin. Immunol. 2008 122 6 1219 -1220
Hauck F., Randriamampita C., Martin E., Gerart S., Lambert N., Lim A., Soulier J., Maciorowski Z., Touzot F., Moshous D., Quartier P., Heritier S., Blanche S., Rieux-Laucat F., Brousse N., Callebaut I., Veillette A., Hivroz C., Fischer A., Latour S., and Picard C. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency J. Allergy. Clin. Immunol. 2012 130 5 1144 -1152 e11
Heimall J., Freeman A., and Holland S.M. Pathogenesis of hyper IgE syndrome Clin. Rev. Allergy. Immunol. 2010 38 1 32 -38
Hoernes M., Seger R., and Reichenbach J. Modern management of primary B-cell immunodeficiencies Pediatr. Allergy. Immunol. 2011 22 8 758 -769
Hori T., Ohnishi H., Teramoto T., Tsubouchi K., Naiki T., Hirose Y., Ohara O., Seishima M., Kaneko H., Fukao T., and Kondo N. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism J. Clin. Immunol. 2012 32 6 1213 -1220
Hsu A.P., Sampaio E.P., Khan J., Calvo K.R., Lemieux J.E., Patel S.Y., Frucht D.M., Vinh D.C., Auth R.D., Freeman A.F., Olivier K.N., Uzel G., Zerbe C.S., Spalding C., Pittaluga S., Raffeld M., Kuhns D.B., Ding L., Paulson M.L., Marciano B.E., Gea-Banacloche J.C., Orange J.S., Cuellar-Rodriguez J., Hickstein D.D., and Holland S.M. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome Blood. 2011 118 10 2653 -2655
Hsu A.P., Sowerwine K.J., Lawrence M.G., Davis J., Henderson C.J., Zarember K.A., Garofalo M., Gallin J.I., Kuhns D.B., Heller T., Milner J.D., Puck J.M., Freeman A.F., and Holland S.M. Intermediate phenotypes in patients with autosomal dominant hyper-IgE syndrome caused by somatic mosaicism J. Allergy. Clin. Immunol. 2013 131 6 1586 -1593
Jain M., Jakubowski A., Cui L., Shi J., Su L., Bauer M., Guan J., Lim C.C., Naito Y., Thompson J.S., Sam F., Ambrose C., Parr M., Crowell T., Lincecum J.M., Wang M.Z., Hsu Y.-M., Zheng T.S., Michaelson J.S., Liao R., and Burkly L.C. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure Circulation. 2009 119 15 2058 -2068
Jefferies J.T.C., Chin A.J., and Jefferies J.L. Barth syndrome Am. J. Med. Genet. C. Semin. Med. Genet. 2013 163C 3 198 -205
Johnson K.D., Hsu A.P., Ryu M.-J., Wang J., Gao X., Boyer M.E., Liu Y., Lee Y., Calvo K.R., Keles S., Zhang J., Holland S.M., and Bresnick E.H. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity J. Clin. Invest. 2012 122 10 3692 -3704
Johnston S.L., Hill S.J., Lock R.J., Dwight J.F., Unsworth D.J., and Gompels M. M. Echocardiographic abnormalities in primary antibody deficiency Postgrad. Med. J. 2004 80 942 214 -218
Jones L.B.K.R., Mcgrogan P., Flood T.J., Gennery A.R., Morton L., Thrasher A., Goldblatt D., Parker L., and Cant A.J. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry Clin. Exp. Immunol. 2008 152 2 211 -218
Jyonouchi S., Mcdonald-Mcginn D.M., Bale S., Zackai E.H., and Sullivan K.E. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features Pediatrics. 2009 123 5 e871 -e877
Kamae C., Nakagawa N., Sato H., Honma K., Mitsuiki N., Ohara O., Kanegane H., Pasic S., Pan-Hammarström Q., Van Zelm M.C., Morio T., Imai K., and Nonoyama S. Common variable immunodeficiency classification by quantifying T-cell receptor and immunoglobulin kappa-deleting recombination excision circles J. Allergy. Clin. Immunol. 2013 131 5 1437 -1440 e5
Kashef S., Moghtaderi M., Borzouee M., and Rezaei N. Giant aneurysm of thoracic and proximal abdominal aorta in a patient with common variable immunodeficiency Iran. J. Allergy. Asthma. Immunol. 2011 10 2 133 -137
Kavanagh D., Spitzer D., Kothari P., Shaikh A., Liszewski M.K., Richards A., and Atkinson J.P. New roles for the major human 3′-5′ exonuclease TREX1 in human disease Cell Cycle. 2008 7 12 1718 -1725
Kawai T. and Malech H.L. WHIM syndrome: congenital immune deficiency disease Curr. Opin. Hematol. 2009 16 1 20 -26
Kazenwadel J., Secker G.A., Liu Y.J., Rosenfeld J.A., Wildin R.S., Cuellar-Rodriguez J., Hsu A.P., Dyack S., Fernandez C.V., Chong C.-E., Babic M., Bardy P.G., Shimamura A., Zhang M.Y., Walsh T., Holland S.M., Hickstein D.D., Horwitz M.S., Hahn C.N., Scott H.S., and Harvey N.L. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature Blood. 2012 119 5 1283 -1291
Kirkpatrick C.H. Chronic mucocutaneous candidiasis Pediatr. Infect. Dis. J. 2001 20 2 197 -206
Kivitie-Kallio S. and Norio R. Cohen syndrome: essential features, natural history, and heterogeneity Am. J. Med. Genet. 2001 102 2 125 -135
Kivitie-Kallio S., Eronen M., Lipsanen-Nyman M., Marttinen E., and Norio R. Cohen syndrome: evaluation of its cardiac, endocrine and radiological features Clin. Genet. 1999 56 1 41 -49
Kobrynski L.J. and Sullivan K.E. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes Lancet. 2007 370 9596 1443 -1452
Kolehmainen J., Black G.C.M., Saarinen A., Chandler K., Clayton-Smith J., Träskelin A.-L., Perveen R., Kivitie-Kallio S., Norio R., Warburg M., Fryns J.-P., Chapelle A.D.L., and Lehesjoki A.-E. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport Am. J. Hum. Genet. 2003 72 6 1359 -1369
Kopel L., Gutierrez P.S., and Lage S.G. Dilated cardiomyopathy in a case of Shwachman–Diamond syndrome Cardiol. Young. 2011 21 5 588 -590
Laufs H., Nigrovic P.A., Schneider L.C., Oettgen H., Del Nido P., Moskowitz I.P.G., Blume E., and Perez-Atayde A.R. Giant cell myocarditis in a 12-year-old girl with common variable immunodeficiency Mayo. Clin. Proc. 2002 77 1 92 -96
Leiding J.W., Marciano B.E., Zerbe C.S., Deravin S.S., Malech H.L., and Holland S.M. Diabetes, renal and cardiovascular disease in p47 phox-/- chronic granulomatous disease J. Clin. Immunol. 2013 33 4 725 -730
Lev A., Amariglio N., Levy Y., Spirer Z., Anikster Y., Rechavi G., Dekel B., and Somech R. Molecular assessment of thymic capacities in patients with Schimke immuno-osseous dysplasia Clin. Immunol. 2009 133 3 375 -381
Liu L., Okada S., Kong X.-F., Kreins A.Y., Cypowyj S., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P., Masson C., Toth B., Flatot J., Migaud M., Chrabieh M., Kochetkov T., Bolze A., Borghesi A., Toulon A., Hiller J., Eyerich S., Eyerich K., Gulacsy V., Chernyshova L., Chernyshov V., Bondarenko A., Maria Cortes Grimaldo R., Blancas-Galicia L., Madrigal Beas I.M., Roesler J., Magdorf K., Engelhard D., Thumerelle C., Burgel P.-R., Hoernes M., Drexel B., Seger R., Kusuma T., Jansson A.F., Sawalle-Belohradsky J., Belohradsky B., Jouanguy E., Bustamante J., Bue M., Karin N., Wildbaum G., Bodemer C., Lortholary O., Fischer A., Blanche S., Al-Muhsen S., Reichenbach J., Kobayashi M., Rosales F.E., Lozano C.T., Kilic S.S., Oleastro M., Etzioni A., Traidl-Hoffmann C., Renner E.D., Abel L., Picard C., Marodi L., Boisson-Dupuis S., Puel A., and Casanova J.-L. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis J. Exp. Med. 2011 208 8 1635 -1648
Loffredo L., Carnevale R., Sanguigni V., Plebani A., Rossi P., Pignata C., De Mattia D., Finocchi A., Martire B., Pietrogrande M.C., Martino S., Gambineri E., Giardino G., Soresina A.R., Martino F., Pignatelli P., and Violi F. Does NADPH oxidase deficiency cause artery dilatation in humans? Antioxid. Redox. Signal. 2013 18 12 1491 -1496
Mahlaoui N., Pellier I., Mignot C., Jais J.-P., Bilhou-Nabera C., Moshous D., Neven B., Picard C., De Saint-Basile G., Cavazzana-Calvo M., Blanche S., and Fischer A. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome Blood. 2013 121 9 1510 -1516
Mandel K., Grunebaum E., and Benson L. Noncompaction of the myocardium associated with Roifman syndrome Cardiol. Young. 2001 11 2 240 -243
Marazzi M.G., Bondi E., Giannattasio A., Strozzi M., and Savioli C. Intracranial aneurysm associated with chronic mucocutaneous candidiasis Eur. J. Pediatr. 2008 167 4 461 -463
Marciano B.E., Rosenzweig S.D., Kleiner D.E., Anderson V.L., Darnell D.N., Anaya-O'brien S., Hilligoss D.M., Malech H.L., Gallin J.I., and Holland S.M. Gastrointestinal involvement in chronic granulomatous disease Pediatrics. 2004 114 2 462 -468
McCarl C.-A., Picard C., Khalil S., Kawasaki T., Röther J., Papolos A., Kutok J., Hivroz C., Ledeist F., Plogmann K., Ehl S., Notheis G., Albert M.H., Belohradsky B.H., Kirschner J., Rao A., Fischer A., and Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy. Clin Immunol. 2009 124 6 1311 -1318 e7
McCluggage W.G., Armstrong D.J., Maxwell R.J., Ellis P.K., and Mccluskey D.R. Systemic vasculitis and aneurysm formation in the Wiskott-Aldrich syndrome J. Clin. Pathol. 1999 52 5 390 -392
McDermott D.H., De Ravin S.S., Jun H.S., Liu Q., Priel D.A.L., Noel P., Takemoto C.M., Ojode T., Paul S.M., Dunsmore K.P., Hilligoss D., Marquesen M., Ulrick J., Kuhns D.B., Chou J.Y., Malech H.L., and Murphy P.M. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis Blood. 2010 116 15 2793 -2802
Mehes K., Kosztolányi G., Kardos M., Horváth M., Opitz J.M., and Reynolds J.F. Cohen syndrome: a connective tissue disorder? Am. J. Med. Genet. 1988 31 1 131 -133
Momma K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome Am. J. Cardiol. 2010 105 11 1617 -1624
Morimoto M., Yu Z., Stenzel P., Clewing J., Najafian B., Mayfield C., Hendson G., Weinkauf J.G., Gormley A.K., Parham D.M., Ponniah U., André J.-L., Asakura Y., Basiratnia M., Bogdanovic R., Bokenkamp A., Bonneau D., Buck A., Charrow J., Cochat P., Cordeiro I., Deschenes G., Fenkçi M., Frange P., Fründ S., Fryssira H., Guillen-Navarro E., Keller K., Kirmani S., Kobelka C., Lamfers P., Levtchenko E., Lewis D.B., Massella L., Mcleod D., Milford D.V., Nobili F., Saraiva J.M., Semerci C., Shoemaker L., Stajic N., Stein A., Taha D., Wand D., Zonana J., Lücke T., and Boerkoel C.F. Reduced elastogenesis: a clue to the arteriosclerosis and emphysematous changes in Schimke immuno-osseous dysplasia? Orphanet. J. Rare. Dis. 2012 7 70
Myers K.C., Bolyard A.A., Otto B., Wong T.E., Jones A.T., Harris R.E., Davies S.M., Dale D.C., and Shimamura A. Variable clinical presentation of Shwachman–Diamond syndrome: update from the North American Shwachman–Diamond Syndrome Registry J. Pediatr. 2014 164 4 866 -870
Nagashima T., Miyanoshita A., Sakiyama Y., Ozaki Y., Stan A.C., and Nagashima K. Cerebral vasculitis in chronic mucocutaneous candidiasis: autopsy case report Neuropathology. 2000 20 4 309 -314
Nahum A., Reid B., Grunebaum E., and Roifman C.M. Matched unrelated bone marrow transplant for Omenn syndrome Immunol. Res. 2009 44 1–3 25 -34
Nahum A., Dadi H., Bates A., and Roifman C.M. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity Autoimmun. Rev. 2012 11 5 341 -347
Nanetti L., Cavalieri S., Pensato V., Erbetta A., Pareyson D., Panzeri M., Zorzi G., Antozzi C., Moroni I., Gellera C., Brusco A., and Mariotti C. SETX mutations are a frequent genetic cause of juvenile and adult onset cerebellar ataxia with neuropathy and elevated serum alpha-fetoprotein Orphanet. J. Rare. Dis. 2013 8 123
Nehme N.T., Schmid J.P., Debeurme F., Andre-Schmutz I., Lim A., Nitschke P., Rieux-Laucat F., Lutz P., Picard C., Mahlaoui N., Fischer A., and De Saint Basile G. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival Blood. 2012 119 15 3458 -3468
Notarangelo L.D. Partial defects of T-cell development associated with poor T-cell function J. Allergy. Clin. Immunol. 2013 131 5 1297 -1305
Ochs H.D., Filipovich A.H., Veys P., Cowan M.J., and Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment Biol. Blood. Marrow. Transpl. 2009 15 1 Suppl 84 -90
Ono M., Goerler H., and Breymann T. Aneurysm of the aortic root in the setting of Wiskott-Aldrich syndrome Cardiol. Young. 2009 19 2 212 -215
Ostergaard P., Simpson M.A., Connell F.C., Steward C.G., Brice G., Woollard W.J., Dafou D., Kilo T., Smithson S., Lunt P., Murday V.A., Hodgson S., Keenan R., Pilz D.T., Martinez-Corral I., Makinen T., Mortimer P.S., Jeffery S., Trembath R.C., and Mansour S. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat. Genet. 2011 43 10 929 -931
Pachlopnik Schmid J. Polymerase epsilon1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”) J. Exp. Med. 2012 209 13 2323 -2330
Pellier I., Girod S.D., Loisel D., Benabidallah S., Proust A., Malhlaoui N., Picard C., Najioullah F., De Saint Basile G., Blanche S., Rialland X., Casanova J.L., and Fischer A. Occurrence of aortic aneurysms in 5 cases of Wiskott-Aldrich syndrome Pediatrics. 2011 127 2 e498 -e504
Perlman S.L., Boder (Deceased) E., Sedgewick R.P., and Gatti R.A. Ataxia-telangiectasia Handb. Clin. Neurol. 2012 103 307 -332
Poliani P.L., Facchetti F., Ravanini M., Gennery A.R., Villa A., Roifman C.M., and Notarangelo L.D. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome Blood. 2009 114 1 105 -108
Puel A., Doffinger R., Natividad A., Chrabieh M., Barcenas-Morales G., Picard C., Cobat A., Ouachee-Chardin M., Toulon A., Bustamante J., Al-Muhsen S., Al-Owain M., Arkwright P.D., Costigan C., Mcconnell V., Cant A.J., Abinun M., Polak M., Bougneres P.-F., Kumararatne D., Marodi L., Nahum A., Roifman C., Blanche S., Fischer A., Bodemer C., Abel L., Lilic D., and Casanova J.-L. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I J. Exp. Med. 2010 207 2 291 -297
Roifman C.M. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: a novel syndrome Clin. Genet. 1999 55 2 103 -109
Roifman C.M., Somech R., Kavadas F., Pires L., Nahum A., Dalal I., and Grunebaum E. Defining combined immunodeficiency J. Allergy. Clin. Immunol. 2012 130 1 177 -183
Roscioli T., Cliffe S.T., Bloch D.B., Bell C.G., Mullan G., Taylor P.J., Sarris M., Wang J., Donald J.A., Kirk E.P., Ziegler J.B., Salzer U., Mcdonald G.B., Wong M., Lindeman R., and Buckley M.F. Mutations in the gene encoding the PML nuclear body protein Sp110 are associated with immunodeficiency and hepatic veno-occlusive disease Nat. Genet. 2006 38 6 620 -622
Sack J. and Friedman E. Cardiac involvement in the Cohen syndrome: a case report Clin. Genet. 1980 17 5 317 -319
Sampaio E.P., Hsu A.P., Pechacek J., Bax H.I., Dias D.L., Paulson M.L., Chandrasekaran P., Rosen L.B., Carvalho D.S., Ding L., Vinh D.C., Browne S.K., Datta S., Milner J.D., Kuhns D.B., Long Priel D.A., Sadat M.A., Shiloh M., De Marco B., Alvares M., Gillman J.W., Ramarathnam V., De La Morena M., Bezrodnik L., Moreira I., Uzel G., Johnson D., Spalding C., Zerbe C.S., Wiley H., Greenberg D.E., Hoover S.E., Rosenzweig S.D., Galgiani J.N., and Holland S.M. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis J. Allergy. Clin. Immunol. 2013 131 6 1624 -1634
Schlame M. and Ren M. Barth syndrome, a human disorder of cardiolipin metabolism FEBS. Lett. 2006 580 23 5450 -5455
Sen S., Wang H., Nghiem C.L., Zhou K., Yau J., Tailor C.S., Irwin M.S., and Dror Y. The ribosome-related protein, SBDS, is critical for normal erythropoiesis Blood. 2011 118 24 6407 -6417
Somech R., Simon A.J., Lev A., Dalal I., Spirer Z., Goldstein I., Nagar M., Amariglio N., Rechavi G., and Roifman C.M. Reduced central tolerance in Omenn syndrome leads to immature self-reactive oligoclonal T cells J. Allergy. Clin. Immunol. 2009 124 4 793 -800
Spencer C.T., Bryant R.M., Day J., Gonzalez I.L., Colan S.D., Thompson W.R., Berthy J., Redfearn S.P., and Byrne B.J. Cardiac and clinical phenotype in Barth syndrome Pediatrics. 2006 118 2 e337 -e346
Su Y. and Swift M. Mortality rates among carriers of ataxia-telangiectasia mutant alleles Ann. Intern. Med. 2000 133 10 770 -778
Tekin M., Dogu F., Taçyildiz N., Akar E., Ikinciogullari A., Ogur G., Yavuz G., Babacan E., and Akar N. 657del5 mutation in the NBS1 gene is associated with Nijmegen breakage syndrome in a Turkish family Clin. Genet. 2002 62 1 84 -88
Toiviainen-Salo S., Pitkänen O., Holmström M., Koikkalainen J., Lötjönen J., Lauerma K., Taskinen M., Savilahti E., Smallhorn J., Mäkitie O., and Kivistö S. Myocardial function in patients with Shwachman-Diamond syndrome: aspects to consider before stem cell transplantation Pediatr. Blood. Cancer. 2008 51 4 461 -467
Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E., Bonilla F.A., Pechacek J., Chandrasekaran P., Browne S.K., Agharahimi A., Gharib A.M., Mannurita S.C., Yim J.J., Gambineri E., Torgerson T., Tran D.Q., Milner J.D., and Holland S.M. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation–polyendocrinopathy–enteropathy–X-linked–like syndrome J. Allergy. Clin. Immunol. 2013 131 6 1611 -1623
Villa A., Notarangelo L.D., and Roifman C.M. Omenn syndrome: inflammation in leaky severe combined immunodeficiency J. Allergy. Clin. Immunol. 2008 122 6 1082 -1086
Villa A., Santagata S., Bozzi F., Giliani S., Frattini A., Imberti L., Gatta L.B., Ochs H. D., Schwarz K., Notarangelo L.D., Vezzoni P., and Spanopoulou E. Partial V(D)J recombination activity leads to Omenn syndrome Cell. 1998 93 5 885 -896
Volkers M., Dolatabadi N., Gude N., Most P., Sussman M.A., and Hassel D. Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish J. Cell. Sci. 2012 125 Pt 2 287 -294
Wang H.-Y., Ma C.A., Zhao Y., Fan X., Zhou Q., Edmonds P., Uzel G., Oliveira J.B., Orange J., and Jain A. Antibody deficiency associated with an inherited autosomal dominant mutation in TWEAK Proc. Natl. Acad. Sci. USA. 2013 110 13 5127 -5132
Weemaes C.M., Van Tol M.J., Wang J., Van Ostaijen-Ten Dam M.M., Van Eggermond M.C., Thijssen P.E., Aytekin C., Brunetti-Pierri N., Van Der Burg M., Graham Davies E., Ferster A., Furthner D., Gimelli G., Gennery A., Kloeckener-Gruissem B., Meyn S., Powell C., Reisli I., Schuetz C., Schulz A., Shugar A., Van Den Elsen P.J., and Van Der Maarel S.M. Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects Eur. J. Hum. Genet. 2013 21 11 1219 -1225
Wilda M., Demuth I., Concannon P., Sperling K., and Hameister H. Expression pattern of the Nijmegen breakage syndrome gene, Nbs1, during murine development Human Molecular Genetics. 2000 9 12 1739 -1744
Writzl K., Cale C.M., Pierce C.M., Wilson L.C., and Hennekam R.C.M. Immunological abnormalities in CHARGE syndrome Eur. J. Med. Genet. 2007 50 5 338 -345
Yalcin A., Aydemir N., Turgut H., Erbay R., and Yalcin A. Common variable immunodeficiency syndrome with right aortic arch: a case report BMC. Infect. Dis. 2004 4 2
Yamamoto S., Yang G., Zablocki D., Liu J., Hong C., Kim S.-J., Soler S., Odashima M., Thaisz J., Yehia G., Molina C.A., Yatani A., Vatner D.E., Vatner S.F., and Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy J. Clin. Invest. 2003 111 10 1463 -1474
Yavuz H. and Chee R. A review on the vascular features of the hyperimmunoglobulin E syndrome Clin. Exp. Immunol. 2010 159 3 238 -244
Zhang J., Quintal L., Atkinson A., Williams B., Grunebaum E., and Roifman C.M. Novel RAG1 mutation in a case of severe combined immunodeficiency Pediatrics. 2005 116 3 e445 -e449
Appendix A1: Cardiac and vascular abnormalities associated with primary immunodeficiency disorders.
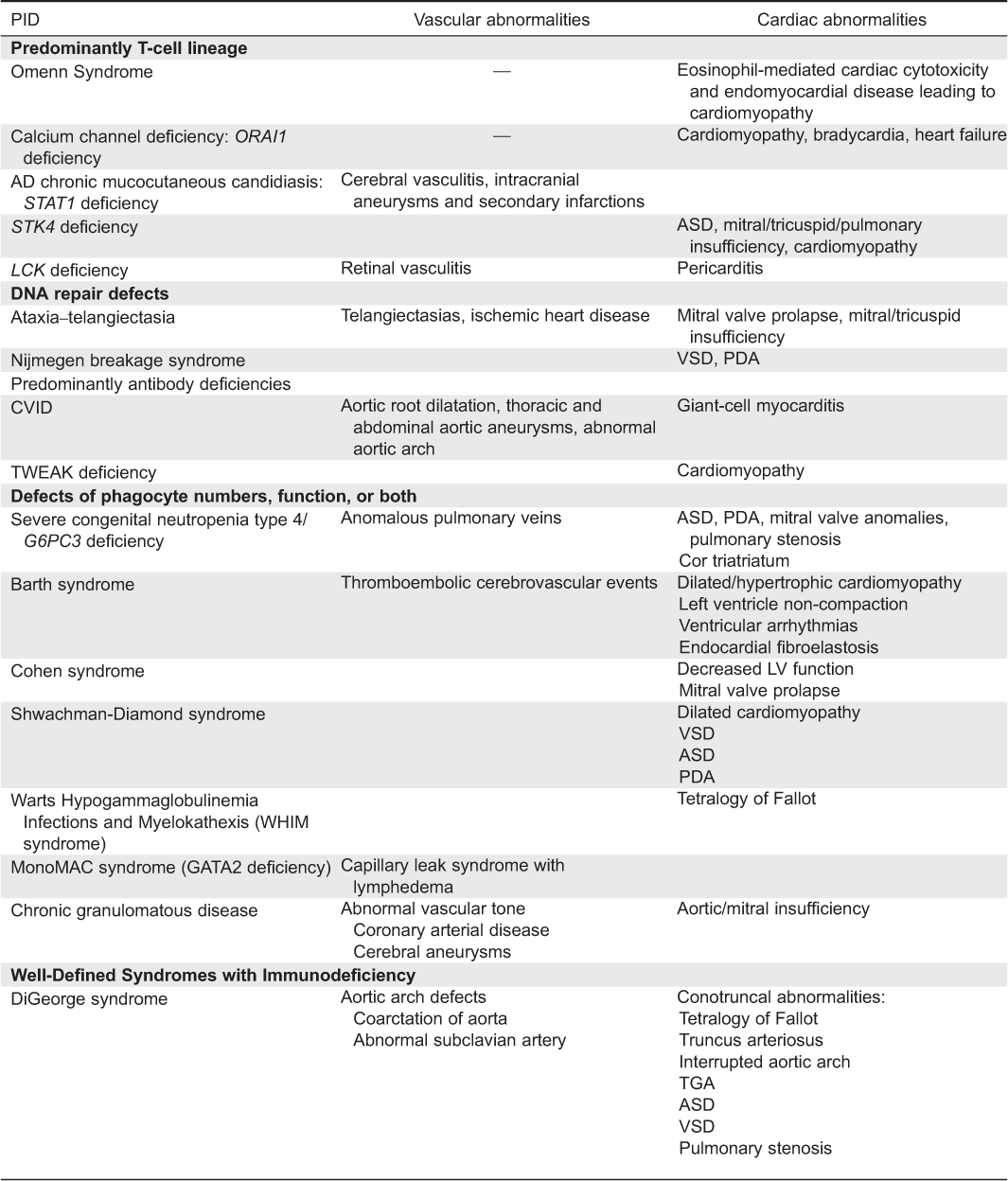
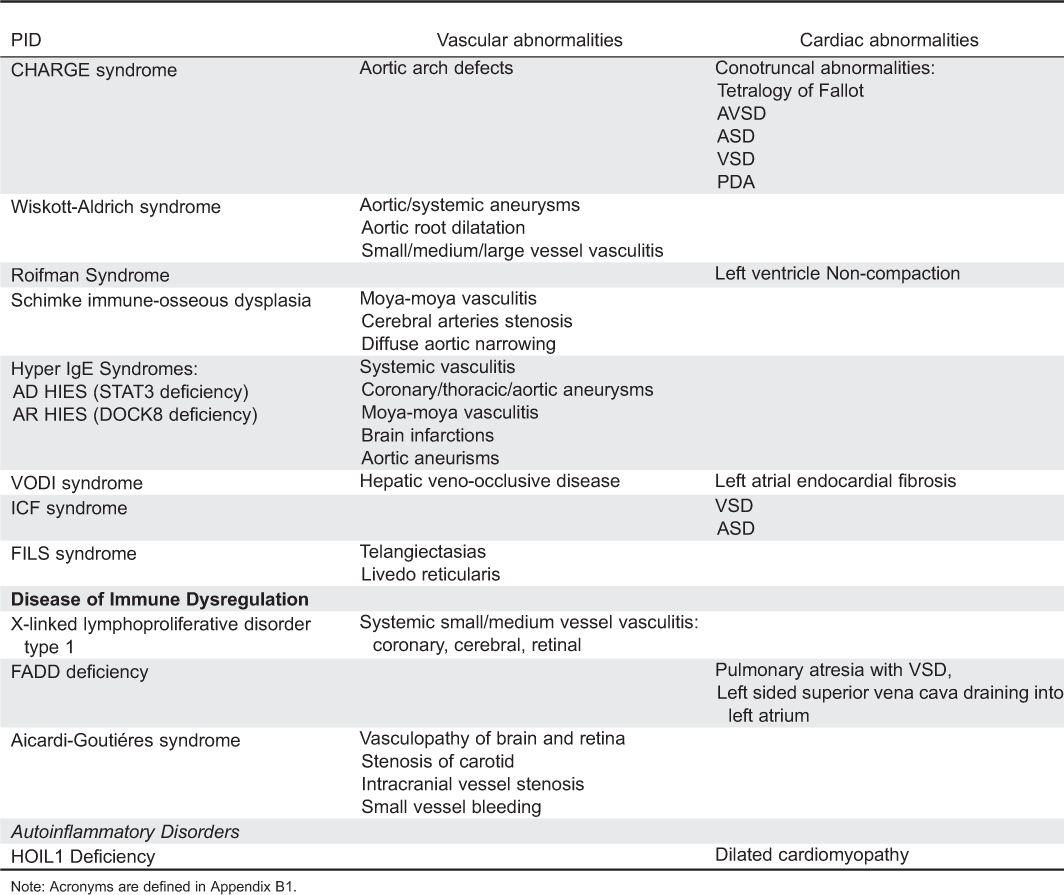
Appendix A2: Primary immunodeficiency disorders associated with cardiac abnormalities.

Appendix A3: Primary immunodeficiency disorders associated with vascular abnormalities.
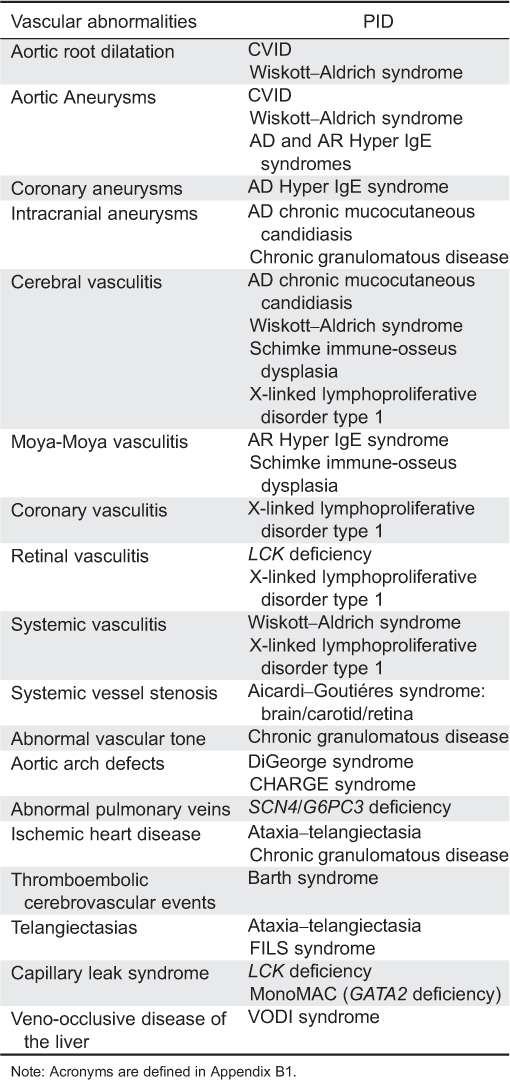
Appendix B1: Acronmyms

Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 3 • September 2015
Pages: 107 - 134
History
Received: 8 September 2014
Accepted: 10 September 2014
Accepted manuscript online: 11 September 2014
Version of record online: 11 September 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Andrea Human, Luis Murguia-Favela, Lee Benson, Idan Roifman, and Eyal Grunebaum. 2015. Cardiovascular abnormalities in primary immunodeficiency diseases. LymphoSign Journal.
2(3): 107-134. https://doi.org/10.14785/lpsn-2014-0013
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
