Whole exome sequencing identifies causative compound heterozygous variants in PRF1 in late-onset familial hemophagocytic lymphohistiocytosis
Abstract
Background: Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening disease in which cells of the immune system are overactivated, leading to uncontrolled inflammation and tissue destruction. Inherited or familial forms of HLH (FHL) are further classified into FHL1 to 5, based on the underlying genetic etiology. The most common form, FHL2, is associated with mutations in the PRF1 gene encoding perforin, a pore-forming glycoprotein required for natural killer and cytotoxic T cell-mediated apoptosis. Importantly, diagnosis of FHL can be challenging, particularly in late-onset cases in which presentation is delayed beyond the first years of life.
Aim: We report the essential role of whole exome sequencing in the diagnostic work-up of a patient with complex, late-onset FHL.
Methods: A comprehensive retrospective chart review was performed.
Results: Our patient presented at 11 years of age with recurrent fever, hepatosplenomegaly, and pancytopenia. In the following years, she was admitted to hospital on multiple occasions, including twice for febrile neutropenia, and once for febrile cytopenia. Serial immune evaluation revealed features of immune dysregulation. While HLH was suspected, she did not fulfil the diagnostic criteria. Initial genetic work-up involving a targeted primary immunodeficiency gene panel identified only a single novel variant of uncertain significance, c.T374C (p.I125T) in PRF1. Subsequently, research-based whole exome sequencing was performed which revealed a second variant, c.C272T (p.A91V), in the same gene. The expanded genetic findings, a set of compound heterozygous missense mutations in PRF1, strengthened the diagnosis of FHL. She later fulfilled the diagnostic criteria for HLH.
Conclusion: Whole exome sequencing identified compound heterozygous mutations in the PRF1 gene in a patient with late-onset FHL.
Statement of Novelty: We report the use of whole exome sequencing to identify compound heterozygous mutations in PRF1, including a novel p.I125T variant not previously identified in FHL.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening disease in which the cells of the immune system are overactivated, leading to uncontrolled inflammation and tissue destruction. Symptoms of HLH include prolonged fever, hepatosplenomegaly, pancytopenia, and hepatitis. Some patients may present with atypical features, including neurological symptoms (ataxia, seizures).
HLH is divided into distinct subgroups, including those with underlying genetic etiology (familial HLH, FHL) or associated with conditions such as malignancy, rheumatologic conditions, immune compromise, or post-immune activation therapy. Still, some forms are not associated with specific conditions. Up to 25% of all HLH cases are inherited, thus, FHL is associated with a positive family history, history of consanguinity, and frequently manifests at an early age. The majority of FHL cases present in infancy (Henter et al. 1991). FHL may also present at a later age, from adolescence through to adulthood (Janka 2012). Secondary forms of HLH are common, and can be triggered by infection, malignancy, and autoimmune disease, appearing as early as the first year of life.
FHL is caused by aberrations in genes involved in the formation or release of cytotoxic granules and is classified into FHL1 to 5. The most common form of FHL is FHL2, which results from mutations in the perforin 1 gene (PRF1). Perforin acts as a membrane-perturbing molecule and is present in natural killer (NK) and cytotoxic T cells. These cells are essential components of both the innate and adaptive immune system, serving to recognize and eliminate pathogen-infected and transformed cells. Central to their function is the ability to release cytotoxic molecules (perforin, granzymes, and granulysin) into target cells. At the cell-cell interface, perforin is needed for the formation of transmembrane pores, allowing trafficking of granzymes into the cytosol of the target cell and triggering death by apoptosis (Abbas 2018). Deficiency of perforin leads to abnormal NK and cytotoxic T cells. Functionally, their failure to eliminate cells such as activated macrophages triggers persistent and uncontrolled inflammation, leading to multi-organ damage. Evidence of hemophagocytosis may also be present but is not pathognomonic.
Patients with late-onset FHL present beyond the first years of life. The exact trigger mechanisms leading to disease remain unknown, however, it has been postulated that certain events (for example, viral infection) can lead to alterations in immune homeostasis and uncontrolled immune activation.
Important challenges which clinicians face in diagnosing HLH include identifying the hemophagocytic syndrome and verifying the underlying genetic etiology. The diagnostic criteria for HLH include fever, splenomegaly, cytopenia in 2 of 3 cell lineages, hypertriglyceridemia with or without hypofibrinogenemia, hemophagocytosis in bone marrow, spleen, lymph nodes or cerebrospinal fluid, reduced NK cell activity, ferritin >500 ng/mL, and soluble CD25 >2400 U/mL. Patients must fulfil at least 5 out of 8 criteria to confirm a diagnosis of HLH (Henter et al. 2007).
Here, we report the case of a patient with late-onset FHL who was suspected of HLH but did not yet fulfil the diagnostic criteria. During her diagnostic odyssey, expanded genetic work-up including whole exome sequencing was essential for the identification of a set of compound heterozygous missense mutations in PRF1, forming the genetic basis of disease in this patient.
Case presentation
Patient
Our patient, currently a 15-year-old female, was born at term to non-consanguineous parents of Eastern European (mother) and Irish (dad) descent. She has a healthy older brother. There is a history of cancer in the paternal grandfather and paternal uncle, but otherwise no history of immunodeficiency or autoimmunity in the family.
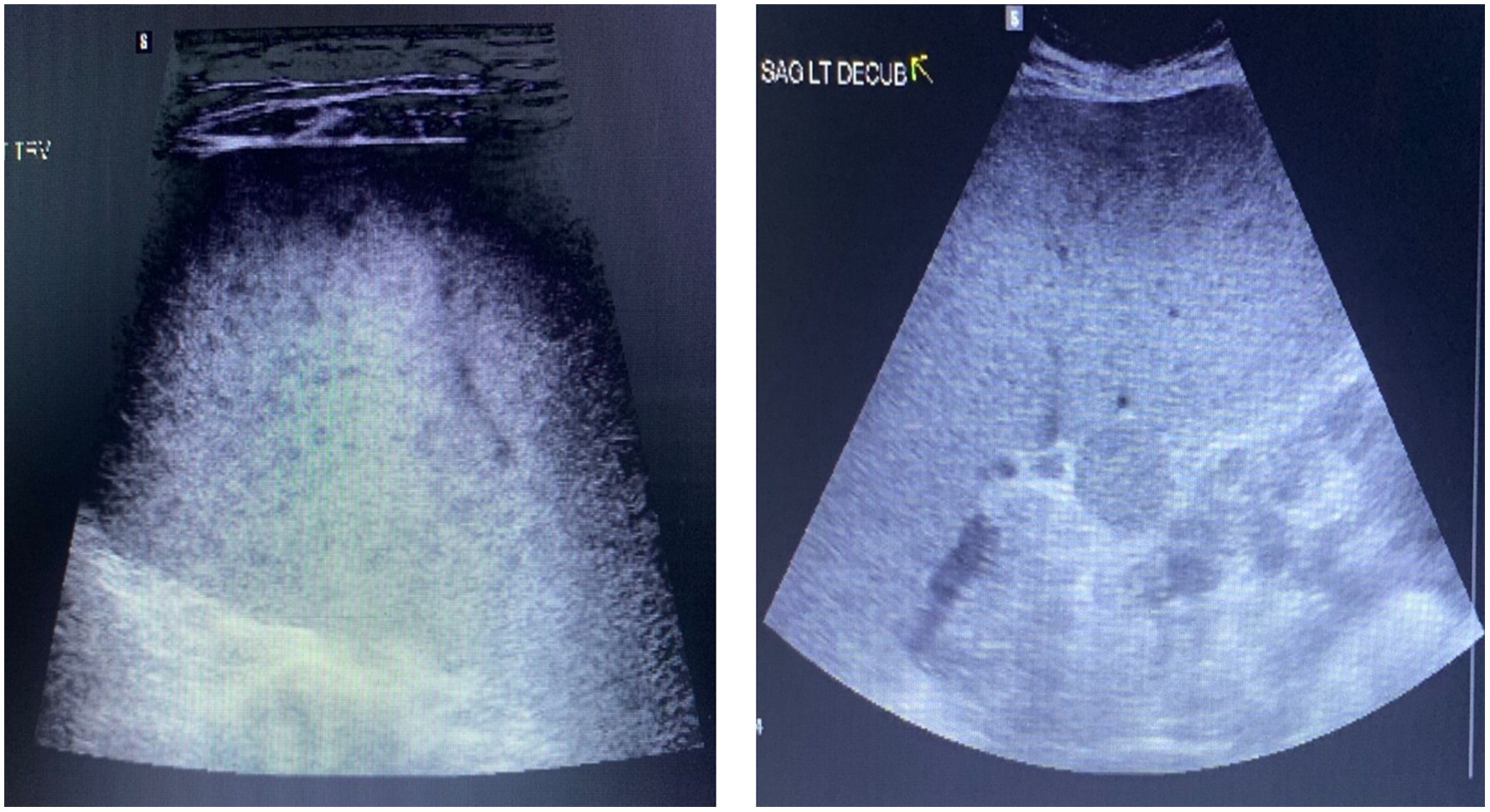
The patient was previously well and first presented at age 11 years with recurrent fevers and pancytopenia, occurring every 4 to 6 months, as well as hepatosplenomegaly. Extensive work-up which included 2 bone marrow aspirates were negative for malignancy or hemophagocytosis. Physical examination revealed massive splenomegaly (20 cm palpable) and her liver was palpable 4 cm below the costal margin. An abdominal ultrasound showed marked splenomegaly with multiple hypoechoic foci throughout the parenchyma (Figure 1). As well, mild hepatomegaly with diffuse slight increased echogenicity of the parenchyma but no focal abnormality was noted. A splenic biopsy revealed focal atypical T lymphoid infiltrate, with abnormal T cell immunophenotype including reduced CD7 and CD45RO expression.
Figure 1:
At age 13 years, she was admitted for severe pneumonia requiring intubation and ventilation. Bilateral lung opacifications and small pleural and pericardial effusions were noted. Laboratory work-up revealed pancytopenia (Table 1). She was treated for PJP pneumonia and given methylprednisolone. Six months later, the patient was again admitted twice for febrile neutropenia. She was administered broad spectrum antibiotics and treated symptomatically for cytopenia. She responded to steroid treatment with resolution of fever and improvement in cell counts and was later started on sirolimus as a steroid-sparing agent. The following month, the patient presented again with history of worsening fever, chill, fatigue, and dry cough. Her laboratory evaluations revealed pancytopenia, elevated ferritin, hypertriglyceridemia, and mildly elevated liver transaminases, for which she was admitted and received stress dosing of hydrocortisone. She received another course of broad-spectrum antibiotics with minimal effect and was later started on anakinra wherein her fever resolved eventually.
Table 1:
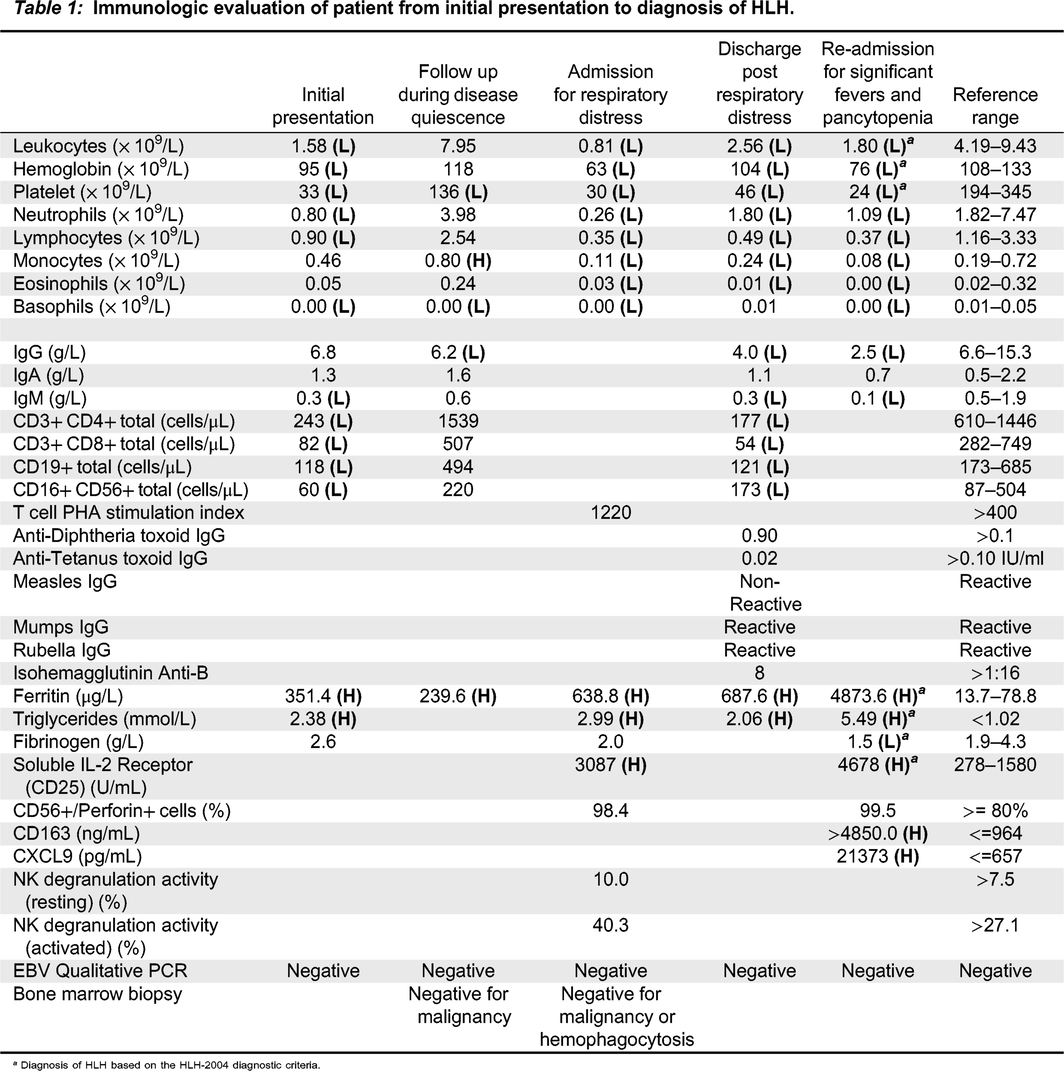
a
Diagnosis of HLH based on the HLH-2004 diagnostic criteria.
Given the remarkable history our patient was evaluated for a possible HLH, however, did not meet the diagnostic criteria. Evaluations of NK cell degranulation and flow cytometric analysis of perforin expression were normal.
Progression of illness
In the following months, our patient developed worsening and persistent fever, pancytopenia with hepatosplenomegaly, and markedly elevated inflammatory markers (Table 1). Her triglyceride levels where elevated at 5.49 mmol/L (normal: <1.02 mmol/L). Infectious disease work-up returned negative, including investigations for cytomegalovirus (CMV), Epstein-Barr Virus (EBV), Human Herpes Virus 6 (HHV6), Herpes Simplex Virus (HSV), Varicella Zoster Virus (VZV), Adenovirus, Parvovirus, Hepatitis B and Hepatitis C virus.
Immune evaluation revealed markedly low cell counts, including low WBC, platelets, neutrophils and lymphocytes. Her CD4, CD8, B cell, and NK cell counts were low (Table 1). Her T cell function was normal, with lymphocyte proliferation responses to the mitogen phytohemagglutinin (PHA) within normal range. While she previously had normal immunoglobulin levels, subsequent evaluation revealed evolving hypogammaglobulinemia and abnormal specific antibody response to tetanus and measles.
Genetic testing
An initial targeted primary immunodeficiency gene panel (Blueprint Genetics) identified a single missense variant of uncertain significance in the PRF1 gene (NM_001083116). The variant in exon 2, c.T374C, results in substitution of isoleucine with threonine at position 125 (p.I125T). This novel variant has not been reported in patients with FHL, however, is predicted in silico to result in loss-of-function. We next performed research whole exome sequencing to identify and (or) rule out other causative variants. Surprisingly, a second variant in PRF1 was detected and subsequently confirmed in a clinical laboratory. The c.C272T missense mutation in exon 2, leading to substitution of alanine with valine at position 91 (p.A91V), was previously reported in association with HLH patients. Thus, the finding of set of compound heterozygous missense mutations in PRF1 confirmed the underlying disease etiology. Sanger segregation revealed the c.T374C (p.I125T) mutation in the patient’s father while her mother harboured the c.272T (p.A91V) variant.
Diagnosis
Our patient eventually fulfilled the HLH criteria (6 out of 8 criteria; fever, splenomegaly, cytopenias, elevated triglyceride and ferritin, and elevated soluble CD25) and was started on HLH treatment protocol (HLH-2004 protocol).
Discussion
We report here the essential role of expansive genetic work-up in a patient with difficult to diagnose, late-onset FHL.
FHL is a rare and life-threatening condition caused by aberrations in genes responsible for cytotoxic granule release and (or) formation. FHL2 results from mutations in the perforin 1 gene (PRF1). Perforin is essential for trafficking of cytotoxic, apoptosis-triggering granzymes into target cells. Structurally, perforin is a 67 kDa protein composed of 3 domains: an N-terminal region that forms the membrane attack complex perforin like/cholesterol dependent cytosylin (MACPF/CDC) domain, an epidermal growth factor (EGF)-like domain, and a calcium dependent C-terminal (C2) domain (Willenbring et al. 2018) (Figure 2). The C-terminal region binds to the lipid membrane in a calcium dependent manner, promoting perforin polymerization and formation of the pore that allows passage of granzymes into target cells. Perforin deficiency results in abnormal NK and cytotoxic T cell function, leading to uncontrolled inflammation and tissue damage in affected individuals.
Figure 2:
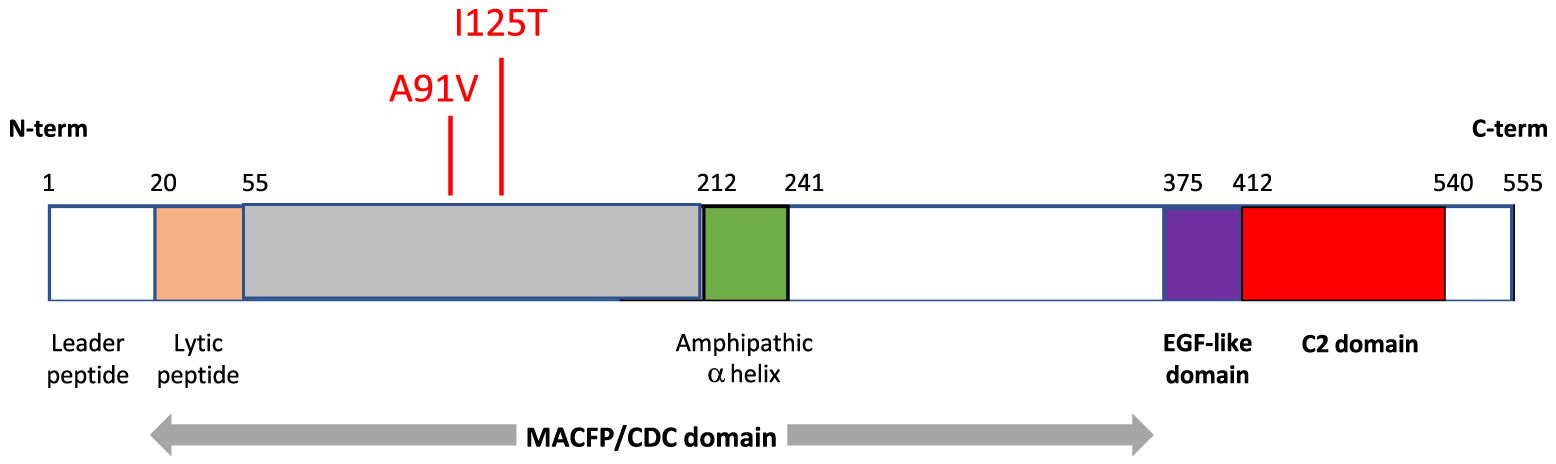
In humans, there are over 460 single nucleotide variants causing silent, frameshift, or missense mutations, in the coding region of PRF1 (Willenbring et al. 2018) (Figure 3). Frequently, when a missense mutation lies within the C2 domain, it tends to be deleterious with the capacity to result in lethality. In contrast, mutations in the MACFP/CDC domain tend to be less deleterious with the exception of those that cause early truncation. FHL2 can result from of single deleterious amino acid substitutions or multiple compound non-deleterious substitutions (Feldmann et al. 2002; Wang 2014).
Figure 3:
The genetic etiology of FHL was first reported in 1999 by Stepp and colleagues (Stepp et al. 1999). Over the years, numerous case reports and studies have been published. A North American study reported 13 novel mutations in PRF1 among 47 patients with HLH (Lee et al. 2004). While Clementi et al. 2002 reported of 2 siblings with mild, adult-onset FHL harbouring compound heterozygous mutations in p.W374X and p.A91V (Clementi et al. 2002). Wang et al. 2014 reported that 18 out of 252 patients with HLH had underlying genetic mutations. Of those, 12 patients had mutations in PRF1. Notably, all 18 patients with HLH had reduced NK cell activity compared to normal controls (Wang 2014).
In our patient, expanded genetic work-up encompassing whole exome sequencing was essential for the identification of a set of compound heterozygous mutations in PRF1. The targeted primary immunodeficiency panel identified a single variant of uncertain significance (c.T374C; p.I125T), but not the second variant (c.C272T; p.A91V), likely affecting the interpretation and impact it would have on protein function. Targeted gene panels sequence pre-defined sets of genes (or regions of interest) that have known or suspected association with disease, producing a small yet manageable data set that provide sufficient depth for identification of rare variants. In contrast, whole exome sequencing provides more expansive coverage, and focuses on coding regions in all exomes that harbour more than 85% of disease variants. Thus, more genetic changes can be identified. The c.T374C (p.I125T) missense variant present in exon 2 has not been previously reported in patients with FHL. In silico tools predict this to result in loss-of-function. The second missense mutation also impacts exon 2, leading to c.C272T (p.A91V) and is associated with decreased perforin protein, misfolding, and reduced NK cytotoxicity. A recent meta-analysis showed that the p.A91V mutation was associated with increased risk of HLH in affected individuals (Zhu et al. 2020). It is likely that this mutation is pathogenic when present in trans with another PRF1 variant, as is the case in our patient. Notably, several studies have reported cases of late-onset FHL in which the patient harboured compound heterozygous mutations in PRF1 gene (Xin-Yi Liu et al. 2021) (Ueda et al. 2007).
Interestingly, evaluations that are often used to support the diagnosis of HLH, including bone marrow examination and perforin protein expression, were normal. Evidence of hemophagocytosis is not always present in HLH and is thus not pathognomonic. Moreover, previous studies have reported that patients with late-onset FHL have near normal perforin function (Lee et al. 2004), perhaps explaining the late presentation. The minimum amount of perforin activity needed to prevent FHL2 remains undefined, however, a murine suggested 10-30% of cytotoxic T cells must express fully active perforin function to prevent FHL2-like symptoms (Terrell and Jordan 2013). Assays for NK cell degranulation, which are frequently crude and unreliable, were also returned normal. Yet, in many cases, it may help to rule out other types of FHL that cause abnormal degranulation (such as FHL3).
Our patient eventually fulfilled 6 out of 8 criteria of the HLH-2004 protocol, including fever, splenomegaly, cytopenias, elevated triglyceride and ferritin, and elevated soluble CD25. She also had elevated inflammatory markers associated with HLH (CD163, CXCL-9, TNF-α).
In sum, we describe a case of difficult to diagnose late-onset FHL in which research whole exome sequencing was essential for identifying set of compound heterozygous mutations in the PRF1 gene. As with many primary immunodeficiencies, it would be important to screen her healthy sibling for the same genetic illness given the possibility of late presentation of FHL2.
REFERENCES
Abbas, A.L. 2018. Cellular and Molecular Immunology Ninth Edition. Elsevier.
Clementi R, Emmi L., Maccario R., Liotta F., Moretta L., Danesino C., and Aricó M. 2002. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood, Sept 100(6): 2266–2267.
Feldmann J., Le Deist F., Ouachée-Chardin M., Certain S., Alexander S., Quartier P., Haddad E., Wulffraat N., Casanova J.L., Blanche S., Fischer A., and de Saint Basile G. 2002. Functional consequences of perforin gene mutations in 22 patients with familial haemophagocytic lymphocytosis. Br J Haematol. 117(4): 965–972.
Henter J.I., Horne A.C., Sabel M., Bryceson Y.T., and Henter J-I. 1991. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Acta Paediatr Scand. 62(2): 346–352.
Henter J.I., Horne A., Aricó M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., McClain K., Webb D., Winiarski J., and Janka G. 2007. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer, 48(2): 124–131.
Janka G. 2012. Familial and Acquired Hemophagocytic Lymphohistiocytosis. Annual Review of Medicine, 166(2): 95–109.
Lee M., Villanueva J., Sumegi J., Zhang K., Kogawa K., Davis J., and Filipovich A.H. 2004. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphocytosis. J. Med. Genet. 41(2): 137–144.
Stepp S.E., Dufourcq-Lagelouse R., Deist F.L., Bhawan S., Certain S., Mathew P.A., Henter J.I., Bennett M., Fischer A., de Saint Basile G., and Kumar V. 1999. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science, 286: 1957–1959.
Terrell, C., and Jordan, M. 2013. Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood, 2618–2621.
Ueda I., Kurokawa Y., Koike K., Ito S., Sakata A., Matsumora T., Fukushima T., Morimoto A., Ishii E., and Imashuku S. 2007. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol, 82(6): 427–442.
Wang Y., Wang Z., Zhang J., Wei Q., Tang R., Qi J., Li L., Ye L., Wang J., and Ye L. 2014. Genetic Features of Late Onset Primary Hemophagocytic in Adolescence or Adulthood. PLoS ONE, 9(9): e107386.
Willenbring R.C., Ikeda Y., Pease L.R., and Johnson A.J. 2018. Human perforin gene variation is geographically distributed. Mol Genet Genomic Med, 6(1): 44–55.
Xin-Yi Liu Y.-B., Nie Y-B., Chen X-J., Gao X-H., Zhai L-J., and Min F-L. 2021. Adult onset type 2 familial hemophagocytic lymphohistiocytosis with PRF1 c.65delC/c.163C>T compound heterozygous mutations: A case report. World J Clin Cases, 9(10): 2289–2295.
Zhu G.H., Zhang L-P., Li Z-G., Wei A., Yang Y., Tian Y., Ma H-H., Wang D., Zhao X-X., Zhao Y-Z., Li N., Liu W., Wang T-Y., and Zhang R. 2020. Associations between PRF1 Ala91Val polymorphism and risk of hemophagocytic lymphohistiocytosis: a meta-analysis based on 1366 subjects. World J Pediatr. 16(6): 598–606.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 9 • Number 4 • December 2022
Pages: 85 - 91
History
Received: 16 November 2022
Accepted: 26 November 2022
Accepted manuscript online: 28 November 2022
Copyright
© 2022.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MarinaSham, RongboZhu, DanieleMerico, and YehonatanPasternak. 2022. Whole exome sequencing identifies causative compound heterozygous variants in PRF1 in late-onset familial hemophagocytic lymphohistiocytosis. LymphoSign Journal.
9(4): 85-91. https://doi.org/10.14785/lymphosign-2022-0014
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Identification of novel compound heterozygous LRBA mutations associated with recurrent hemophagocytic lymphohistiocytosis and CNS manifestations
