Abstracts from the Immunodeficiency Canada—9th SCID Symposium, 28 October 2021
Griscelli syndrome type 2 treated with hematopoietic stem cell transplantation using an unrelated cord blood donor
Adil Adatia1, Vicky Breakey2, Jennifer Mackenzie3, Yogi Chopra4, Rae Brager5
1Division of Clinical Immunology and Allergy, Department of Medicine, McMaster University, Hamilton, ON, Canada
2Division of Pediatric Hematology and Oncology, Department of Pediatrics, McMaster Children's Hospital, McMaster University, Hamilton, ON, Canada
3Division of Genetics, Department of Pediatrics, McMaster Children's Hospital, McMaster University, Hamilton, ON, Canada
4Division of Haematology/Oncology, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
5Division of Rheumatology, Immunology, and Allergy, Department of Pediatrics, McMaster Children's Hospital, McMaster University, Hamilton, ON, Canada
Introduction: Primary hemophagocytic lymphohistiocytosis (HLH) is caused by inborn errors of immunity that impair cytotoxic lymphocyte activity leading to systemic hyperinflammation (Sieni et al. 2014). Griscelli Syndrome type 2 (GS2) is a rare condition associated with oculocutaneous albinism and HLH due to biallelic loss-of-function mutations in RAB27A (Menasche et al. 2000). RAB27A encodes the GTPase Rab27a, which facilitates docking of cytotoxic granules on to the plasma membrane in NK and cytotoxic T cells in preparation for exocytosis of granule proteins into a target cell (Neeft et al. 2005). Partial albinism occurs in GS2 as Rab27a is similarly involved in melanosome docking to actin during their transport from melanocytes to keratinocytes (Kuroda et al. 2003). Microscopic examination of the hair demonstrates characteristic pigment clumping, which may aid in the diagnosis in addition to gene sequencing and assays of NK cell degranulation. Treatment of GS2 involves management of HLH using standard protocols and urgent hematopoietic stem cell transplant (HSCT) (Sieni et al. 2014).
Clinical Features: A 33-day-old male presented to a community hospital with a history of lethargy, decreased feeding, and fever. The patient was found to have bicytopenia (Hb 71 g/L and platelets 59 × 109/L) and an abdominal mass and was thus transferred to McMaster Children’s Hospital for further evaluation.
He had no relevant medical history in the first month of life. He was conceived by in vitro fertilization with intracytoplasmic sperm injection (ICSI) and was born at term after a healthy pregnancy by spontaneous vaginal delivery with no post-natal complications. His parents were of Indian and Dutch origins, respectively, and were non-consanguineous. They had one healthy five-year old daughter.
On examination, he was febrile (38.9 degrees Celsius) and tachycardic (180 bpm). He appeared pale but was alert and active. His hair had a slight silver hue, which the parents confirmed was more prominent at birth. There were no dysmorphic features and no rashes or bruising. He had splenomegaly on abdominal examination. His cardiopulmonary, musculoskeletal, and neurologic exams were unremarkable. The patient was resuscitated with intravenous crystalloid and one transfusion of packed red blood cells, and he was started on empiric broad spectrum antibiotics for possible sepsis.
During the admission, his platelet count dropped to a nadir of 17 × 109/L and he was found to have highly elevated ferritin (3220 μg/L), triglycerides (2.91 mmol/L), and c-reactive protein (253 mg/L). No source of infection was identified by cerebral spinal fluid, blood, or urine cultures. His nasopharyngeal swab for respiratory viruses, serology for parvovirus b19, and PCR for CMV and EBV were negative. No consolidation was seen on chest radiograph. Bone marrow biopsy showed erythroid hyperplasia and reactive lymphocytes with no evidence of a myeloproliferative or lymphoproliferative disorder and no hemophagocytosis. The hair sample submitted for microscopy was not processed due to laboratory error. The patient improved spontaneously over several days and was discharged.
Exome sequencing of 14 genes associated with HLH was obtained, which revealed a heterozygous change in RAB27A (c.239+1G>T) affecting the canonical splice site of exon 3. This variant was considered disease-causing given a previous report of a patient with GS2 who was homozygous for this variant (Panigrahi et al. 2015), in-silico analysis (GeneSplicer, NNSplice, and MaxEntScan) predicted the variant to be likely pathogenic, and it has a low allele frequency in population controls (0.0016% in gnomAD). A chromosomal microarray subsequently demonstrated loss of material from the long arm of chromosome 15 approximately 461 kb in size with deletion of exons 4-6 of RAB27A (15q21.3(55061156_55522407)). The diagnosis of GS2 was confirmed by familial segregation studies showing that the mother was heterozygous for the splice site mutation and the father was heterozygous for the deletion, and by the patient’s NK cell degranulation assay, which showed only 14% NK cell degranulation upon cell activation.
The patient subsequently required multiple admissions for intermittent fever and anemia requiring transfusion, and at age 11 weeks he was readmitted for pancytopenia and fever. His ferritin and D-dimer were highly elevated at 6700 μg/L and 1000 μg/L, respectively, and ALT was elevated at 161 U/L. He was diagnosed with HLH and treated with dexamethasone 10 mg/m2 daily and one dose of etoposide 150 mg/m2 intravenously, which improved his blood counts and hyperferritinemia.
The patient was referred to our local transplant centre for HSCT. The patient’s elder sister was not an HLA match, so an unrelated cord blood unit (UCB) mismatched at two alleles (6/8) was selected. He received conditioning with treosulfan, fludarabine, cyclophosphamide and anti-thymocyte globulin and tacrolimus and methylprednisolone for graft-versus-host disease (GvHD) prophylaxis. His course was complicated by oral mucositis requiring intravenous morphine for pain management. At day 137 post-transplant, he was well with no evidence of GvHD and consistent full donor chimerism.
Discussion: We report a patient with GS2-related HLH due to compound heterozygosity for a deleterious variant in RAB27A affecting the splice site of exon 3, and a 461 kb deletion on chromosome 15, who successfully underwent HSCT using a 6/8 UCB. The largest series of HSCT in GS2 reported 35 consecutive patients from a single center (Al-Mofareh et al. 2020). The most important predictor of survival post-transplant was HLH prior to HSCT; survival was 100% in those transplanted prior to the development of HLH compared with 53.3% in those transplanted after HLH onset. Therefore, early diagnosis and initiation of management is critically important. Delayed diagnosis, however, is common and in this case was due to barriers in obtaining and the slow turnaround time of genetic tests, loss of the hair sample, and intermittent spontaneous improvement of the patient, which confounded the clinical picture. This case highlights the need for broader and more rapid access to genetic testing for the investigation of inborn errors of immunity, consistent with the findings of a recent Canadian study (Branch et al. 2021).
Accurate diagnosis of primary HLH requires genetic testing, and next generation sequencing technologies are widely employed for the diagnosis of GS2 (Castano-Jaramillo et al. 2021). Though useful for the detection of numerous variant types, analysis of whole exome data using established copy number variant algorithms may miss a substantial minority of deleterious structural variants (Fromer et al. 2012; Miyatake et al. 2015). Microarray-based methods for detecting structural variants are thus important complementary diagnostic tools, as illustrated in this case. In addition, ICSI infants, such as the present child, have higher rates of chromosomal aberrations that are either inherited (and potentially the cause parental infertility) or de novo due to the procedure (Bonduelle et al. 2002). A high index of suspicion for pathologic structural variants in these children is thus warranted.
The case series of HSCT in GS2 discussed above (Al-Mofareh et al. 2020) reported 14 patients who received mismatched UCB (7/8 match in 6 patients and 6/8 in 8 patients). Acute GvHD and graft failure were more common in patients who received UCB compared to those who received matched-related donors (43% vs 16% and 21.4% vs. 5.3%, respectively), but mortality was similar in both groups (approximately 36%). HSCT using UCB for these patients therefore appears to be a reasonable option in the absence of a readily available matched donor, especially given the urgency of HSCT and the toxicities associated with chemotherapy used for treatment or prevention HLH while a wider donor search is conducted.
Novel therapeutic strategies for primary HLH may change the treatment approach for GS2 prior to HSCT as the specific pathways driving hyperinflammation in these patients is better understood. Locatelli et al. (2020) reported the efficacy of the anti-interferon gamma monoclonal antibody emapalumab in 34 patients with primary HLH (five of whom had GS2) and found a favorable overall response rate of 65%, leading to its regulatory approval in the U.S.A. Clinical trials of the janus kinase inhibitor ruxolitinib in primary HLH are also ongoing (e.g., NCT04120090, NCT04551131).
Conclusion: We present a patient with GS2 caused by a deletion in chromosome 15 affecting exons 4-6 and a deleterious change in the exon 3 splice site, both in RAB27A. We highlight (1) the need for more rapid access to genetic testing for the investigation of inborn errors of immunity to reduce diagnostic delays; (2) the importance of careful investigation for pathogenic copy number variants in children with suspected primary HLH for timely diagnosis and treatment initiation, especially in ISCI neonates; and (3) the successful use of a 6/8 UCB for HSCT in GS2, which adds to existing evidence for UCB transplant when matched related donors are not available.
REFERENCES
Al-Mofareh, M., Ayas, M., Al-Seraihy, A., Siddiqui, K., Al-Jefri, A., Ghemlas, I., Alsaedi, H., El-Solh, H., Al-Sweedan, S., Al-Saud, B., Al-Mousa, H., Al-Dhekri, H., Arnaout, R., Mohammed, R., Al-Muhsen, S., and Al-Ahmari, A. 2020. Hematopoietic stem cell transplantation in children with Griscelli syndrome type 2: a single-center report on 35 patients. Bone Marrow Trans. 55(10): 2026–2034.
Bonduelle, M., Van Assche, E., Joris, H., Keymolen, K., Devroey, P., Van Steirteghem, A., and Liebaers, I. 2002. Prenatal testing in ICSI pregnancies: incidence of chromosomal anomalies in 1586 karyotypes and relation to sperm parameters. Hum. Rep. 17(10): 2600–2614.
Branch, A., Modi, B., Bahrani, B., Hildebrand, K.J., Cameron, S.B., Junker, A.K., Turvey, S.E., and Biggs, C.M. 2021. Diverse clinical features and diagnostic delay in monogenic inborn errors of immunity: A call for access to genetic testing. Pediatr. Aller. Immun.
Castano-Jaramillo, L.M., Lugo-Reyes, S.O., Cruz Munoz, M.E., Scheffler-Mendoza, S.C., Duran McKinster, C., Yamazaki-Nakashimada, M.A., Espinosa-Padilla, S.E., and Saez-de-Ocariz Gutierrez, M.D.M. 2021. Diagnostic and therapeutic caveats in Griscelli syndrome. Scand. J. Immunol. 93(6): e13034.
Fromer, M., Moran, J.L., Chambert, K., Banks, E., Bergen, S.E., Ruderfer, D.M., Handsaker, R.E., McCarroll, S.A., O’Donovan, M.C., Owen, M.J., Kirov, G., Sullivan, P.F., Hultman, C.M., Sklar, P., and Purcell, S.M. 2012. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 91(4): 597–607.
Kuroda, T.S., Ariga, H., and Fukuda, M. 2003. The actin-binding domain of Slac2-a/melanophilin is required for melanosome distribution in melanocytes. Mol. Cell. Biol. 23(15): 5245–5255.
Locatelli, F., Jordan, M.B., Allen, C., Cesaro, S., Rizzari, C., Rao, A., Degar, B., Garrington, T.P., Sevilla, J., Putti, M.C., Fagioli, F., Ahlmann, M., Dapena Diaz, J.L., Henry, M., De Benedetti, F., Grom, A., Lapeyre, G., Jacqmin, P., Ballabio, M., and de Min, C. 2020. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J. Med. 382(19): 1811–1822.
Menasche, G., Pastural, E., Feldmann, J., Certain, S., Ersoy, F., Dupuis, S., Wulffraat, N., Bianchi, D., Fischer, A., Le Deist, F., and de Saint Basile, G. 2000. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 25(2): 173–176.
Miyatake, S., Koshimizu, E., Fujita, A., Fukai, R., Imagawa, E., Ohba, C., Kuki, I., Nukui, M., Araki, A., Makita, Y., Ogata, T., Nakashima, M., Tsurusaki, Y., Miyake, N., Saitsu, H., and Matsumoto, N. 2015. Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an 'exome-first' approach. J. Hum. Genet. 60(4): 175–182.
Neeft, M., Wieffer, M., de Jong, A.S., Negroiu, G., Metz, C.H., van Loon, A., Griffith, J., Krijgsveld, J., Wulffraat, N., Koch, H., Heck, A.J., Brose, N., Kleijmeer, M., and van der Sluijs, P. 2005. Munc13–4 is an effector of rab27a and controls secretion of lysosomes in hematopoietic cells. Mol. Biol. Cell. 16(2): 731–741.
Panigrahi, I., Suthar, R., Rawat, A., and Behera, B. 2015. Seizure as the presenting manifestation in Griscelli syndrome type 2. Pediatr. Neurol. 52(5): 535–538.
Sieni, E., Cetica, V., Hackmann, Y., Coniglio, M.L., Da Ros, M., Ciambotti, B., Pende, D., Griffiths, G., and Arico, M. 2014. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. 5: 167.
Compound heterozygosity with a novel missense variant in PRF1 in a patient with hemophagocytic lymphohistiocytosis (HLH)
Rongbo Zhu1, Chaim Roifman2,3
1Division of Clinical Immunology and Allergy, Department of Medicine, Schulich School of Medicine and Dentistry, London, ON, Canada
2Division of Clinical Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
3The Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and the University of Toronto, Toronto, ON, Canada
Background: Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening condition characterized by uncontrolled hyperinflammatory with evidence of T-cell and NK cell dysfunction (George 2014). Most common causes of HLH are secondary in nature and have been implicated in many conditions including infection, malignancy and autoimmunity (George 2014). However, primary or familial HLH (FHL) should be suspected in children or adolescence presenting in hyperinflammatory states as FHL is invariably fatal unless prompt treatment is implemented with bone marrow transplant as a curative intent (Cetica et al. 2016; Chinn et al. 2018). Monogenic causes have been implicated in FHL with mutations in PRF1 accounting for 20–40% of all FHL cases (Gholam et al. 2011). We describe a case of HLH caused by 2 separate missense variants of PRF1 in a compound heterozygous fashion.
Methods: Clinical and laboratory information was collected by retrospective chart review.
Results:
Clinical Features at Presentation: This is a 14-year-old previously healthy Caucasian female born to non-consanguineous parents who initially presented at 11 years of age with recurrent fevers occurring every 4-6 months lasting 3-5 days with associated fatigue but no other infectious symptoms. She was found to have evidence of splenomegaly and lymphadenopathy with new onset pancytopenia at the age of 12. A bone marrow biopsy performed at the time showed adequate cellularity with no evidence of malignant transformations or hemophagocytosis.
Her pancytopenia continued to decline and at age 13, she presented to hospital with significant respiratory distress. A BAL was not performed given her instability and she was treated with presumed Pneumocystis jirovecii pneumonia (PJP) pneumonia. She then had 2 further admissions of febrile neutropenia within the next 4 months from her recurrent fevers and associated pancytopenia.
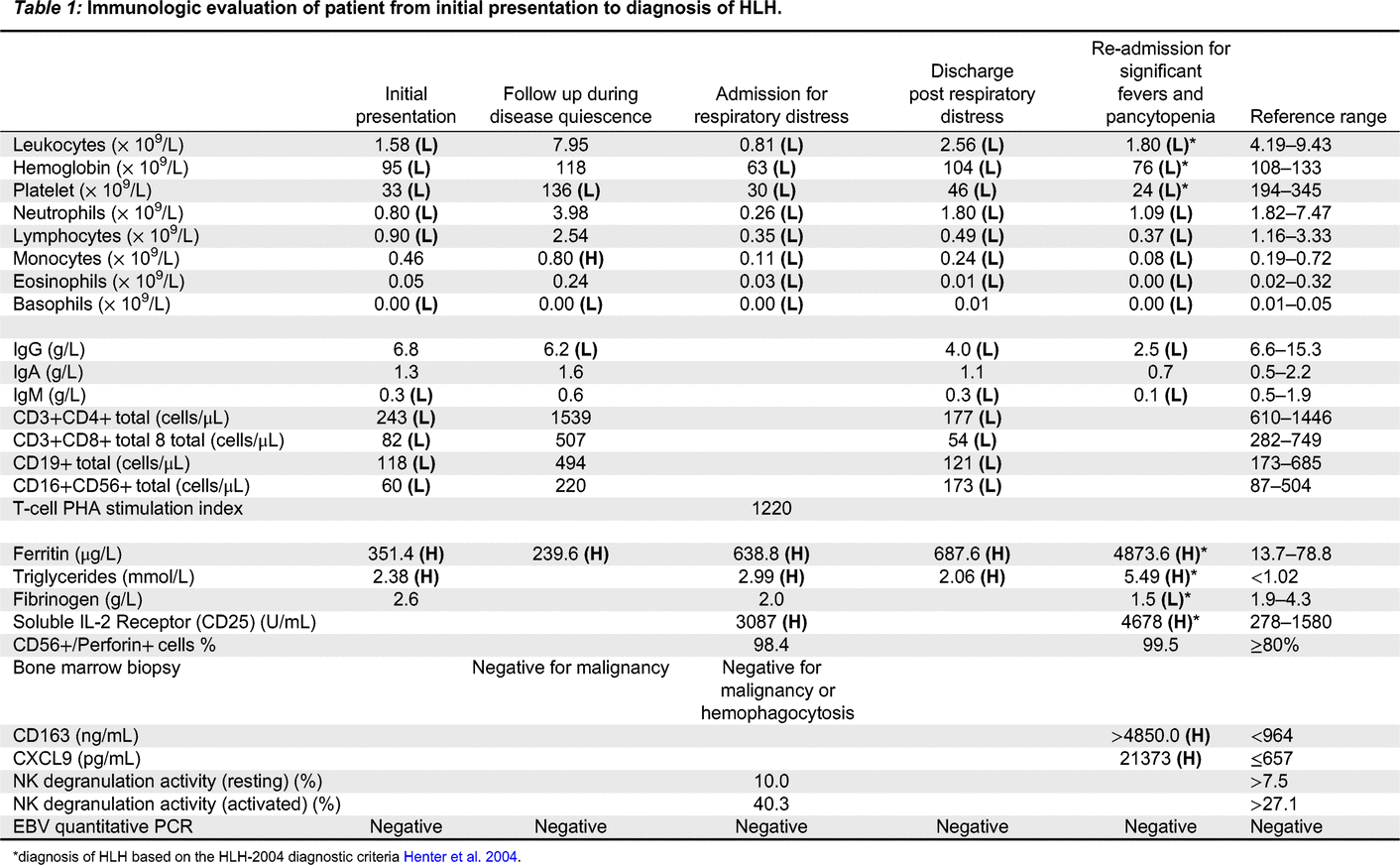
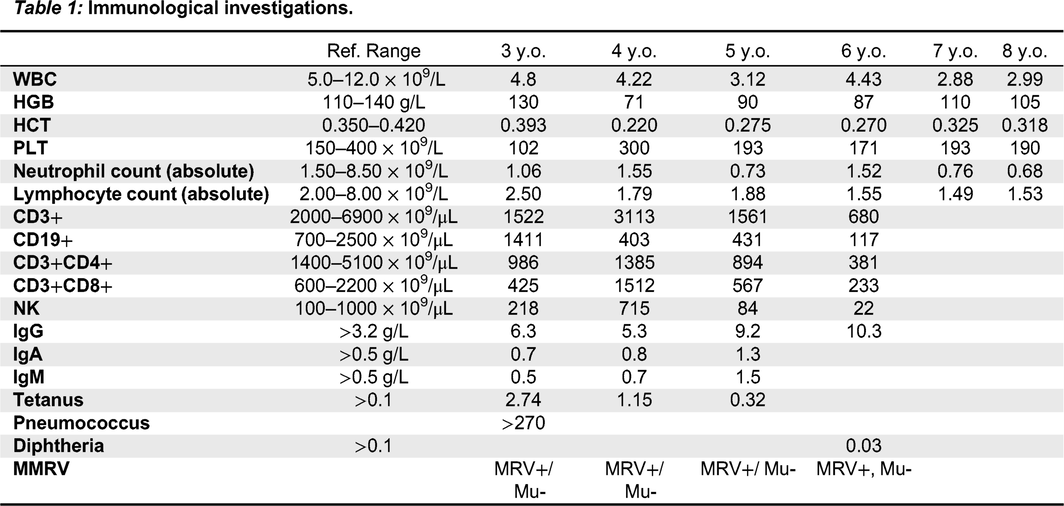
Immunologic features: Her relevant blood work from initial presentation to fulminant HLH is highlighted in Table 1.
Table 1:
*
diagnosis of HLH based on the HLH-2004 diagnostic criteria Henter et al. 2004.
She underwent genetic testing with a PID Panel from BluePrint that identified a novel heterozygous missense mutation in PRF1 c.374T>C p.(Ile125Thr) that has not been reported in literature to cause disease. A subsequent whole exome sequencing (WES) confirmed the initial novel Ile125Thr (I125T) variant but also a second heterozygous missense mutation in PRF1 c.273C>T p.(Ala91Val).
Throughout her admissions, she was seen by multiple specialists including Infectious Disease who ruled out an infectious etiology. Despite her inflammation, she did not initially meet criteria for HLH and given her persistent and worsening pancytopenia and recurrent fevers in the absence of infection or malignancy, she was empirically started on high dose prednisone with good effect. She was then transitioned to sirolimus as a steroid sparing agent but had another episode of breakthrough fevers and pancytopenia during steroid weaning requiring admission. She was then started on anakinra and continued steroid therapy. Despite these therapies, the patient continued to have significant pancytopenia and evidence of elevated inflammatory markers which ultimately lead to the diagnosis of HLH based on the 2004-HLH diagnostic criteria (Table 1; Henter et al. 2007). She has since started treatment for HLH and is currently undergoing an allogenic bone marrow transplant as definitive management.
Discussion: Mutations of the PRF1 gene have been implicated in FHL (Lee et al. 2004). Reports of the PRF1 variant Ala91Val (A91V) has had conflicting results in the literature as up to 9% of this allele was found in the healthy population (zur Stadt et al. 2004). However, perforin expression and activity in A91V was found to be significantly reduced compared to wildtype protein suggesting a possible predisposition for HLH in these individuals, especially if a second non-functional variant of PRF1 was inherited (Voskoboinik et al. 2007). Cases of HLH have been reported in patients with A91V variants with a second variant causing a nonsense mutation (Trp374Stop) in the PRF1 gene (Clementi et al. 2002).
There have been no reports in the literature regarding disease associated with the I125T variant of the PRF1 gene. However, a different missense mutation in the same codon PRF1 c.374T>A p.(Ile125Asn) was associated with a patient with T-cell lymphoma (Ding and Yang 2013). We postulate the missense A91V variant along with the novel missense I125T variant together acted as loss of function in a compound heterozygous way to cause HLH in this patient.
Conclusion: We report a patient with HLH due to compound heterozygosity from a missense A91V variant and a novel I125T missense variant in the PRF1 gene.
REFERENCES
Cetica, V., Sieni, E., Pende, D., Danesino, C., Fusco, C., and De Locatelli, F. 2016. Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry. J. Allergy Clin. Immunol. 137(1): 188. Available from /pmc/articles/PMC4699615/.
Chinn, I.K., Eckstein, O.S., Peckham-Gregory, E.C., Goldberg, B.R., Forbes, L.R., Nicholas, S.K., Mace, E.M., Vogel, T.P., Abhyankar, H.A., Diaz, M.I., Heslop, H.E., Krance, R.A., Martinez, C.A., Nguyen, T.C., Bashir, D.A., Goldman, J.R., Stray-Pedersen, A., Pedroza, L.A., Cecilia Poli, M., Aldave-Becerra, J.C., McGhee, S.A., Al-Herz, W., Chamdin, A., Coban-Akdemir, Z.H., Jhangiani, S.N., Muzny, D.M., Cao, T.N., Hong, D.N., Gibbs, R.A., Lupski, J.R., Orange, J.S., McClain, K.L., and Allen, C.E. 2018. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. 132(1): 89. Available from /pmc/articles/PMC6034641/.
Clementi, R., Emmi, L., Maccario, R., Liotta, F., Moretta, L., and Danesino, C. 2002. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 100(6): 2266–2267.
Ding, Q., and Yang, L.-Y. 2013. Perforin gene mutations in 77 Chinese patients with lymphomas. World J. Emerg. Med. 4(2): 128. Available from https://pubmed.ncbi.nlm.nih.gov/25215106/.
George, M.R. 2014. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J. Blood. Med. 5: 69. Available from /pmc/articles/PMC4062561/.
Gholam, C., Grigoriadou, S., Gilmour, K.C., and Gaspar, H.B. 2011. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin. Exp. Immunol. 163(3): 271. Available from /pmc/articles/PMC3048610/.
Henter, J.I., Horne, A.C., Aricó, M., Egeler, R.M., Filipovich, A.H., and Imashuku, S. 2007. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer. 48(2): 124–131.
Lee, S.M., Villanueva, J., Sumegi, J., Zhang, K., Kogawa, K., and Davis, J. 2004. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J. Med. Genet. 41(2): 137–144. Available from https://jmg.bmj.com/content/41/2/137.
Voskoboinik, I., Sutton, V.R., Ciccone, A., House, C.M., Chia, J., and Darcy, P.K. 2007. Perforin activity and immune homeostasis: the common A91V polymorphism in perforin results in both presynaptic and postsynaptic defects in function. Blood. 110(4): 1184–1190.
zur Stadt, U., Beutel, K., Weber, B., Kabisch, H., Schneppenheim, R., and Janka, G. 2004. A91V is a polymorphism in the perforin gene not causative of an FHLH phenotype. Blood. 104(6): 1909–1910. Available from http://ashpublications.org/blood/article-pdf/104/6/1909/1701815/zh801804001907b.pdf.
Virally triggered multisystem inflammatory disease associated with a pathogenic mutation in a novel gene.
Lisa Liang1, Marianne Miguel1, Patrick Frosk2,3, Janet Chou4, Abduarahman Almutairi4, Craig Platt4, Raif Geha4, Tamar Rubin1
1Division of Clinical Immunology and Allergy, Department of Pediatrics, University of Manitoba, Winnipeg, Canada
2Division of Biochemistry and Medical Genetics, Department of Pediatrics, University of Manitoba, Winnipeg, Canada
3Children’s Hospital Research Institute of Manitoba, Winnipeg, Canada
4Division of Clinical Immunology and Allergy, Boston Children’s Hospital, Harvard Medical School, Boston, United States
Background: Next generation sequencing technologies have greatly accelerated diagnosis and treatment of inborn errors of immunity or primary immunodeficiencies (PIDs) (Stray-Pederson et al. 2017; Tangye et al. 2020). However, despite application of clinically validated immunologic and genetic tests, including broad PID gene panels and clinical whole exome sequencing, many individual patients with suspected PIDs remain without a genetic diagnosis or targeted treatment (Heimall et al. 2018).
Methods: Consenting patients and their parents from the pediatric immunodeficiency clinic at the Children’s Hospital of Winnipeg with suspected PID, who have undergone standard of care clinical investigations, and yet remain without a clinical diagnosis are frequently enrolled in local and international research studies, with the aim of obtaining a genetic diagnosis.
After obtaining consent from our patient’s parents, we enrolled our patient in a pan-Canadian study (Care4Rare Solve). In addition, simultaneously, after consent from the parents, her clinical case summary, variant call files (VCFs) and skin biopsy for fibroblast cell lines were obtained and sent to colleagues with a research focus on genetic diagnosis of monogenic PIDs, at an outside center. Genematcher was used to connect investigators with interest in the common candidate gene, and functional testing was performed to determine the impact of the mutation on RNA and protein expression and immune function.
Results:
Clinical Features: A now 4 year-old female was born to consanguineous parents of Oji-Cree First Nations descent. She presented with three severe multisystem inflammatory HLH-like episodes. Identified infectious triggers for these episodes included Group A streptococcus, vaccine-strain Measles, Ebstein-Barr virus (EBV), Influenza B and Staphylococcus aureus. In addition to these HLH-like episodes, our patient had an umbilical stump infection at 10 days of life (positive for Streptococcus anginosus), and recurrent episodes of mucocutaneous thrush.
Her first major illness occurred at 5 months old, and consisted of fever, extremity rash and anorexia, rapidly progressing to lethargy, labored breathing and respiratory failure. At the time, a chest x-ray revealed pneumonia, and blood culture was positive for Group A streptococcus. She had leukocytosis (WBC 50 × 109/L), thrombocytopenia (platelets 42 × 109/L), anemia, transaminitis (AST and ALT peaked at 9000 U/L), hyperferritinemia (peak of 12 000 ug/L), rhabdomyolysis, splenomegaly, acute kidney injury and acute respiratory distress syndrome (ARDS). During her acute illness, she was noted to have mildly decreased B-cell and NK cell counts. Her presentation was most consistent with toxic shock syndrome with sepsis and multi-organ failure. She was treated in hospital for 3 weeks, and made a good recovery. She remained well from 6 to 11 months of age, although she was noted to have mild global developmental delay.
At 12 months of age, our patient presented with her second HLH-like illness. A week after receiving her second set of vaccinations (DTaP-IPV-Hib, MMR and Men-C vaccines), she developed fever, cough and rhinorrhea. This quickly progressed to respiratory failure again, with ARDS and multi-organ dysfunction. She was noted to have hepatosplenomegaly, rhabdomyolysis and rash. Investigations revealed leukocytosis (WBC >50 × 109/L), anemia (hemoglobin 75 g/L), thrombocytopenia (platelets 20 × 109/L), transaminitis (AST 281 U/L, ALT 108 U/L), hyperferritinemia (peak 2300 ug/L) and elevated CRP (70 mg/L). She was found to have vaccine-strain measles virus via PCR of the nasopharyngeal aspirate, and measles virus IgM in her serum. A low level of EBV was also found via PCR in her blood (6.6 × 102 copies/mL). A bone marrow aspirate showed a hypercellular marrow and few hemophagocytes, and liver biopsy revealed non-specific inflammation, with negative staining for EBV, CMV, and HSV. She was treated with ribavirin, vitamin A, methylprednisone (1 mg/kg), and a dose of IVIG. She remained in the intensive care unit for six weeks.
After the second episode, she appeared to recover well clinically. However, upon follow-up over the next 20 months, she was found to have persistent EBV viremia, transaminitis, and cirrhosis. Investigations of the liver disease included multiple biopsies and imaging studies, and were negative for autoimmune and infectious causes. The etiology of the liver disease was suspected to be bystander damage due to the ongoing underlying immune dysregulation issue. Workup for hematopoietic stem cell transplant (HSCT) was initiated, but the lack of diagnosis contributed to reluctance to perform this risky and invasive procedure.
At 33 months of age, our patient presented with progressive dyspnea, lethargy and fever that quickly deteriorated to respiratory failure. Her nasopharyngeal aspirate by positive for Influenza B, and ETT cultures were positive for staphylococcus aureus. As in her two previous episodes, she developed severe ARDS, bicytopenia, marked leukocytosis, transaminitis, hyperferritinemia (peak 2546 ug/L), splenomegaly and acute kidney injury. She was intubated for 3 weeks. Unfortunately, during this episode she also developed a new profound neurologic injury, and seizure activity. An MRI of her brain revealed bilateral diffuse restriction in her cerebrum, cerebellum, thalami and dura. She was treated with oseltamivir and antibiotics. She also received higher dose IVIG (1 g/kg), and, because HLH was suspected, she was initially treated with high dose dexamethasone. HSCT was considered again, but not further pursued because of her severely impaired neurologic status.
In terms of management, the patient was advised against receiving further vaccinations (particularly live vaccines), and was placed on TMP-SMX prophylaxis (discontinued by the family). Antifungal prophylaxis was considered but not pursued due to the patient’s liver disease. At the current time, the patient is alive, on antiepileptic therapy, and receiving weekly subcutaneous replacement doses of IVIG (0.5 g/kg/month). Our patient is also being considered for Janus kinase inhibitor, and HSCT.
Immunological Features: Initial immune investigations were performed after recovery from her first multisystem inflammatory episode (after having received her first two sets of vaccines, and before receiving any IVIG). This revealed normalized WBC and differential, hemoglobin and platelets, and normal levels of IgG, IgA, IgM, and IgE. At this time, she also had normalized T cell, B cell and NK cell counts (B cell and NK cell counts were found to be low during the initial presentation), although she had a mildly low percentage of T cells expressing the recent thymic emigrant marker CD31 (42.4%, median 67% for age), and a mildly low percentage of CD4 T cells expressive the naïve marker RA (51%; median 89%). She had demonstrated protective titers to tetanus and diphtheria, and detectable IgG against streptococcus pneumoniae. Total hemolytic complement (CH50) was normal, and imaging revealed the presence of a normal spleen.
Follow up testing after her second inflammatory episode revealed IgG to measles mumps and rubella (after having received MMR vaccine and IVIG), a normal mitogen proliferative response to PHA, PWM and ConA, normal neutrophil oxidative burst index, and normal expression of CD18/CD11abc and CD15s. HIV testing was negative. She had normal NK cell cytotoxicity and CD107a expression. She had a decreased percentage of NK cells expressing perforin, which was thought to be related to the relative expansion of the CD56 bright NK cell population at the time of testing. She also had increased MCF of granzyme B in NK cells. Of note, she had ongoing EBV positivity in her blood by PCR at this time.
Mutation Analysis: Initial genetic testing for this patient consisted of an HLH/periodic fever gene panel (SickKids, negative), chromosome microarray (London Health Sciences Center; 11% regions of homozygosity but otherwise not diagnostic for a monogenic immune deficiency), and trio whole exome sequencing (GeneDx). Targeted testing of the known Northern Cree IKBKB homozygous duplication mutation was also performed, and this was negative.
Re-analysis of the exome sequencing was performed, with sequencing and variant annotation, variant prioritization and classification, and transcriptomics profiling (Vavassori et al. 2021). Tissue expression, fibroblast stimulation assay, Western blotting and other functional testing are also described in detail in this paper.
After research-based re-analysis of the patient’s clinical exome, a homozygous mutation with a high CADD score was identified in ZNFX1 which is a novel gene associated with impaired immunity in mice. Through enrollment in Care4Rare Solve, additional patients with mutations in this same gene were identified, resulting in ongoing international collaborative study and functional validation of the mutation (Heimall et al. 2018). ZNFX1 is an important regulator of the response to double-stranded nucleic acids stimuli following viral infections. Patients with ZNFX1 deficiency are susceptible to severe viral infections and a multisystem inflammatory disease. The result of this collaboration is the discovery of a new monogenic PID, enhanced understanding of antiviral immunity in humans, and the possibility of targeted treatment like Janus kinase inhibitor and hematopoietic stem cell transplantation (HSCT) (Vavassori et al. 2021).
Conclusions: For patients in whom there is a high clinical suspicion of a monogenic PID, but no diagnosis despite optimal clinical-based immunologic and genetic testing, research-based testing, and, in many cases, collaboration with experts at local and outside centers is essential to optimize diagnosis, treatment, and prognostication for these unique patients. Although enhanced investigations such as whole genome sequencing may be required in some cases, in other cases, such as this one, re-analysis of existing genetic investigations resulted in accurate diagnosis. Furthermore, collaboration through Care4Rare Solve allowed for the identification of other patients with mutations in the same gene, and the possibility of targeted treatments for our patient, who is currently undergoing evaluation for stem cell transplantation, and for whom Janus kinase inhibition may be considered for management of future HLH-like episodes (Vavassori et al. 2021).
REFERENCES
Heimall, J.R., Hagin, D., and Hajjar, J. 2018. Use of Genetic Testing for Primary Immunodeficiency Patients [published correction appears in J. Clin. Immunol. 2018 May 21]. 38(3): 320–329. doi: 10.1007/s10875-018-0489-8. Available from http://care4rare.ca/discovery.
Stray-Pederson, A., Sorte, H.S., and Samarakoon, P. 2017. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J. Allergy Clin. Immunol. 139(1): 232–245.
Tangye, S.G., Al-Herz, W., and Bousfiha, A. 2020. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 40(1): 24–64. doi: 10.1007/s10875-019-00737-x.
Vavassori, S., Chou, J. Faletti, L.E., Haunerdinger, V., Opitz, L., Joset, P., Fraser, C.J., Prader, S., Gao, X., Schuch, L.A., Wagner, M., Hoefele, J., Maccari, M.E., Zhu, Y., Elakis, G., Gabbett, M.T., Forstner, M., Omran, H., Kaiser, T., Kessler, C., Olbrich, H., Frosk, P., Almutairi, A., Platt, C.D., Elkins, M., Weeks, S., Rubin, T., Planas, R., Marchetti, T., Koovely, D., Klämbt, V., Soliman, N.A., von Hardenberg, S., Klemann, C., Baumann, U., Lenz, D., Klein-Franke, A., Schwemmle, M., Huber, M., Sturm, E., Hartleif, S., Häffner, K., Gimpel, C., Brotschi, B., Laube, G., Güngör, T., Buckley, M.F., Kottke, R., Staufner, C., Hildebrandt, F., Reu-Hofer, S., Moll, S., Weber, A., Kaur, H., Ehl, S., Hiller, S., Geha, R., Roscioli, T., Griese, M., and Schmid, J.P. 2021. Multisystem inflammation and susceptibility to viral infections in human ZNFX1 deficiency. J. Allergy Clin. Immunol. S0091-6749(21): 00613-8. doi: 10.1016/j.jaci.2021.03.045. Epub ahead of print. PMID: 33872655.
Atopy and Autoimmunity in Roifman Syndrome
Jenny Garkaby1, Jessica Willett Pachul1, Brenda Reid1,2, Chaim M. Roifman1,2
1Division of Immunology & Allergy, Department of Pediatrics, Hospital for Sick Children and University of Toronto, Toronto, ON
2Canadian Centre for Primary Immunodeficiency, Hospital for Sick Children, Toronto, ON
Introduction: Roifman Syndrome was first described as a novel association of humoral deficiency, spondyloepiphyseal dysplasia, retinal dystrophy, poor pre- and postnatal growth, developmental delay and facial dysmorphism (Roifman 1999). Because the initial published cases of Roifman Syndrome were males it had been suggested that Roifman Syndrome may be an X-linked disorder (Gray et al. 2011), however it was 16 years later that the underlying genetic etiology was revealed (Merico et al. 2015). Roifman Syndrome is a rare autosomal recessive multi-system disorder caused by mutations that disrupt highly conserved positions of the RNU4ATAC small nuclear RNA gene (Merico et al. 2015). Roifman Syndrome is characterized by immune deficiency, mainly humoral, spondyloepiphyseal dysplasia, retinal dystrophy, growth retardation and developmental delay (Roifman 1999; Roifman and Melamed 2003; Merico et al. 2015; Dinur Schejter et al. 2016, 2017).
The association of atopy and immune dysregulation is well described in some of the inborn errors of immunity (IEI) such as Common Variable Immune Deficiency (CVID), Wiskott-Aldrich syndrome (WAS), Hyper-IgE syndromes (HIES), Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX), lipopolysaccharide-responsive beige-like anchor (LRBA) deficiency and other IEI (Castagnoli et al. 2021). The most common atopic manifestation is atopic dermatitis (Castagnoli et al. 2021), however, other immune dysregulation features have been reported and include Immune thrombocytopenia (ITP), autoimmune hemolytic anemia (AIHA), enteropathy, thyroiditis, food allergy, allergic rhinitis etc.
To date, only some of the reported RS patients were described as having atopy and autoimmunity. This report aims to delineate the atopic features as well as immune dysregulation in Roifman Syndrome patients.
Methods: Patients were identified if they were diagnosed with Roifman Syndrome at the Hospital for Sick Children between 1997 and 2020. Data compiled included demographics, clinical manifestations with a focus on atopy and autoimmunity, immunological evaluation, and molecular diagnosis of Roifman Syndrome. Laboratory evaluation included complete blood count and differential, eosinophilic count, lymphocyte subsets, total immunoglobulins including IgE, specific vaccine responses, and T-cell stimulation responses to mitogen and antigen, thyroid function tests, and relevant imaging.
Results: Overall, of the 17 patients, 15 patients (88%) were identified to have atopic, or autoimmune manifestations as shown in Table 1. Atopic dermatitis was observed frequently, of the 17 patients, 14 had atopic dermatitis. The onset of eczema was at early infancy for all affected patients with different clinical outcomes, ranging from mild to severe eczema. All were managed with topical medications and no immune modulation therapy was required. Two patients were identified to have environmental allergies in the form of allergic rhinitis and conjunctivitis. Furthermore, 7 out of 17 patients (41%) had clinical features of immune dysregulation. The most common feature of autoimmunity in Roifman Syndrome patients was identified as autoimmune hypothyroidism. Hypothyroidism was observed in 5 patients (29%), with transient disease in one of the five patients. Cytopenias were also observed in 5 patients as follows: AIHA in two patients, ITP in two patients, neutropenia in two patients.
Table 1:
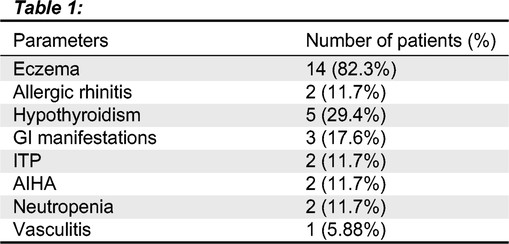
Gastrointestinal involvement was seen in three patients. One patient was diagnosed with moderate chronic eosinophilic esophagitis and distal rectum proctitis which resolved, one patient had evidence of micro-colitis on endoscopy and one with recurrent mouth sores with no other gastrointestinal manifestations. None of the patients in our cohort had chronic active arthritis except for one patient who was diagnosed with Gout’s arthritis. One patient sustained an episode of CNS vasculitis, presenting as seizures, requiring treatment with steroids, Cyclophosphamide, Mycophenolate mofetil and high dose of IV immunoglobulins. Only 2 patients remained free of atopy or autoimmunity.
With regards to clinical outcome of atopy, to date, atopic dermatitis has not resolved for any of the patients. Allergic rhinitis was managed successfully with Antihistamines and local steroid spray. There were no noted endocrine complications related to hypothyroidism. None of the patients remained on immunomodulatory treatment following the initial presentation. One patient remained on G-CSF therapy for neutropenia.
Laboratory evaluation related to atopy showed normal IgE levels for all patients, including those with eczema. Only 3 of the 17 patients had mild eosinophilia ranging between 500-970 cells/μL.
Discussion: Roifman Syndrome is a rare multi-system genetic disorder characterized by humoral deficiency, growth retardation, spondyloepiphyseal dysplasia, retinal dystrophy, and developmental delay, while atopy and autoimmunity were rarely described thus far as a key feature in Roifman Syndrome (Roifman 1999; Robertson et al. 2000; Roifman and Melamed 2003; de Vries et al. 2006; Fairchild et al. 2011; Merico et al. 2015; Dinur Schejter et al. 2016; Bogaert et al. 2017). IEI are a heterogeneous group of disorders, the number of genetic defects associated with atopy and immune dysregulation is rapidly growing. In this report, we retrospectively evaluated the atopic features and autoimmunity of 17 patients with Roifman Syndrome. Most of the patients presented with eczema since early infancy. The prevalence of atopic dermatitis in our study is significantly higher to that found in the general population, 82% and 22.6% respectively (Bylund et al. 2020). Hypothyroidism was also frequently observed. Other autoimmune manifestations included AIHA, ITP, neutropenia, gastrointestinal involvement with colitis or eosinophilic infiltration and CNS vasculitis. Our results showed that atopy is a significant feature among Roifman Syndrome patients. The laboratory evaluation of IgE and eosinophilia were not remarkable in this study with only few cases of mild eosinophilia and normal IgE levels in all the patients.
We have demonstrated that both atopy and autoimmunity contribute to the clinical picture of Roifman Syndrome patients. We suggest that screening for atopy should be a part of the clinical evaluation in Roifman Syndrome patients. Furthermore, for Roifman Syndrome patients we suggest that regular and life-long follow up should be performed in all cases for screening periodically, not only for changes in immune function, but also for autoimmune manifestations such as hypothyroidism, ITP, AIHA, neutropenia as well as gastrointestinal evaluation.
To conclude, our current study expands the spectrum of the clinical features in Roifman Syndrome patients as well as IEI presenting with atopy and autoimmunity.
REFERENCES
Bogaert, D.J., Dullaers, M., Kuehn, H.S., Leroy, B.P., Niemela, J.E., Wilde, H.D., Schryver, S.D., Bruyne, M.D., Coppieters, F., Lambrecht, B.N., Baets, F.D., Rosenzweig, S.D., Baere, E.D., and Haerynck, F. 2017. Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Scientific Rep. 7(1): 1–12. doi: 10.1038/s41598-017-02434-4.
Bylund, S., von Kobyletzki, L.B., Svalstedt, M., and Svensson, Å. 2020. Prevalence and incidence of atopic dermatitis: A systematic review. Acta Dermato-Venereologica, 100(100-year theme Atopic dermatitis): 320–329. doi: 10.2340/00015555-3510.
Castagnoli, R., Lougaris, V., Giardino, G., Volpi, S., Leonardi, L., La Torre, F., Federici, S., Corrente, S., Cinicola, B.L., Soresina, A., Cancrini, C., Marseglia, G.L., Cardinale, F., and Immunology Task Force of the Italian Society of Pediatric Allergy and Immunology (SIAIP). 2021. Inborn errors of immunity with atopic phenotypes: A practical guide for allergists. World Allergy Org. J. 14(2): 100513. doi: 10.1016/j.waojou.2021.100513.
Dinur Schejter, Y. Merico, D., Manson, D., Reid, B., and Vong L. 2016. A Novel Mutation in Roifman Syndrome Redefines the Boundaries of the Sm Protein-Binding Site. LymphoSign J. 3(November): 159–163. doi: 10.14785/lymphosign-2016-0015.
Dinur Schejter, Y., Merico, D., Manson, D., Reid, B., and Vong, L. 2017. A homozygous mutation in the stem II domain of RNU4ATAC causes typical Roifman syndrome. npj Genomic Med. 2(1): 1–7. doi: 10.1038/s41525-017-0024-5.
Fairchild, H.R., Fairchild, G., Tierney, F., McCartney, D.L., Cross, J.J., and de Vries, P.J. 2011. Partial agenesis of the corpus callosum, hippocampal atrophy, and stable intellectual disability associated with Roifman syndrome. Am. J. Med. Gene. Part A. 155(10): 2560–2565. doi: 10.1002/ajmg.a.34215.
Gray, P.E.A., Sillence, D., and Kakakios, A. 2011. Is Roifman syndrome an X-linked ciliopathy with humoral immunodeficiency? Evidence from 2 new cases. Inter. J. Immunogene. 38(6): 501–505. doi: 10.1111/j.1744-313X.2011.01041.x.
Merico, D., Roifman, M., Braunschweig, U., Yuen, R.K.C., Alexandrova, R., Bates, A., Reid, B., Nalpathamkalam, T., Wang, Z., Thiruvahindrapuram, B., Gray, P., Kakakios, A., Peake, J., Hogarth, S., Manson, D., Buncic, R., Pereira, S.L., Herbrick, J.-A., Blencowe, B.J., Roifman, C.M., and Scherer, S.W. 2015. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman Syndrome by disrupting minor intron splicing. Nat. Commun. 6. doi: 10.1038/ncomms9718.
Robertson, S.P., Rodda, C., and Bankier, A. 2000. Hypogonadotrophic hypogonadism in Roifman syndrome. Clinical Genet. 57(6): 435–438. doi: 10.1034/j.1399-0004.2000.570606.x.
Roifman, C.M. 1999. Antibody deficiency, growth retardation, spondyloepiphyseal dysplasia and retinal dystrophy: A novel syndrome. Clinical Genet. 55(2): 103–109. doi: 10.1034/j.1399-0004.1999.550206.x.
Roifman, C.M., and Melamed, I. 2003. A novel syndrome of combined immunodeficiency, autoimmunity and spondylometaphyseal dysplasia. Clinical Genet. 63(6): 522–529. doi: 10.1034/j.1399-0004.2003.00033.x.
de Vries, P.J., McCartney, D.L., McCartney, E., Woolf, D., and Wozencroft, D. 2006. The cognitive and behavioural phenotype of Roifman syndrome. J. Intellect. Disab. Res. 50(9): 690–696. doi: 10.1111/j.1365-2788.2006.00817.x.
Haploinsufficiency of A20 in a patient with 6q23.2-23.3 deletion
Keely Loewen1, Lily Siok Hoon Lim2, Tamar Rubin1
1Department of Pediatrics & Child Health, University of Manitoba, Section of Clinical Immunology and Allergy, Winnipeg, MN, Canada
2Department of Pediatrics & Child Health, University of Manitoba, Section of Pediatric Rheumatology, Winnipeg, MN, Canada
Background: Haploinsufficiency of A20 (HA20) is an autoinflammatory condition characterized by early onset recurrent fevers, mucosal ulcers, ocular inflammation, and arthritis. The disorder was first described in 2016 (Zhou et al. 2016), and has sometimes been compared to Behcet’s disease, another autoinflammatory syndrome also characterized by recurrent ulcers (Aeschlimann et al. 2018). Although initially described in individuals with heterozygous germline mutations in TNFAIP3, HA20 has subsequently been described in patients with chromosomal deletions, specifically on chromosome 6 (Franco-Jarava et al. 2018). Both patients with TNFAIP3 mutations (single nucleotide variants and small insertions/deletions) (Aeschlimann et al. 2018), and patients with larger chromosome 6q deletions have been described tohave recurrent sinopulmonary infections, although the cause of this predisposition is not well characterized (Dutrannoy et al. 2009).
We report on a patient with developmental delay, hearing loss, short stature, recurrent sinopulmonary infections since early childhood, recurrent fevers, and recurrent oral and genital ulcers, diagnosed with heterozygous 6q23.2-23.3 deletion prior to the first reported description of HA20. Although she initially had a generally reassuring immune workup and elusive diagnosis, upon reassessment years later, she was found to have clinical features consistent with HA20, confirmed by her known 6q chromosomal deletion encompassing the TNFAIP3 gene. To our knowledge, this is the 8th case of HA20 caused by a contiguous chromosome 6q deletion, and the second case in the literature caused specifically by 6q23.2.-23.3 deletion. We present a detailed immune assessment for our patient.
Case Presentation: Our patient was born at 34 weeks’ gestation due to pre-eclampsia, to non-consanguineous parents of European background. She had a past medical history of mild global developmental delay, behavioural concerns, and recurrent oto-sino-pulmonary infections since 18 months of age. She received >40 courses of oral antibiotics as well as several courses of intravenous antibiotics for primarily respiratory tract infections. She also required tympanostomy tubes for effusions, inserted 9 times due to extrusion. She had a history of chronic nasal congestion and sensorineural hearing loss.
Diagnostic workup included sweat chloride testing (negative), video fluoroscopic swallowing study (normal), aeroallergen skin testing (negative), FeNO for assessment of ciliary dyskinesia (normal), and 2 ciliary biopsies for electron microscopy assessment. One of the biopsies showed ciliary abnormalities in 25% of the specimen, which was inconclusive in the setting of inflammation. CT scan did not show evidence of bronchiectasis, but did show parenchymal scarring.
At age 2, she began to experience episodes of 1–3 oral ulcers, every 3 months. These would resolve spontaneously, without scarring. She had gingivitis and poor dentition. Recurrent fevers occurred between the ages of 6 and 10, with episodes lasting 2 to 4 days, and no specific pattern. There was no associated pharyngitis, lymphadenopathy, arthritis, arthralgias or skin rashes. Recurrent genital ulcers began at age 12, lasting two weeks at a time.
Workup of her developmental delay included a chromosomal microarray in 2015, around age 6, which identified a de novo chromosome 6q deletion (6q23.2q23.3). Based on known features of this microdeletion at the time, she underwent a sleep study (showing central sleep apnea), as well as cardiology assessment (normal). She is pending an MRI brain to rule out CNS vasculitis, as she also was experiencing headaches.
Updated immune workup showed persistently elevated ESR, intermittent normocytic anemia, increased proportion of transitional B cells at 15.2% of B cells (normal 1.7–7.4%), and decreased proportion of CD21+CD19+ B cells at 89.1% (normal 94.5–99.8%). Notably, the proportion of memory and class-switched memory B cells was normal, and response to pneumococcal polysaccharide immunization was robust.
Table 1:
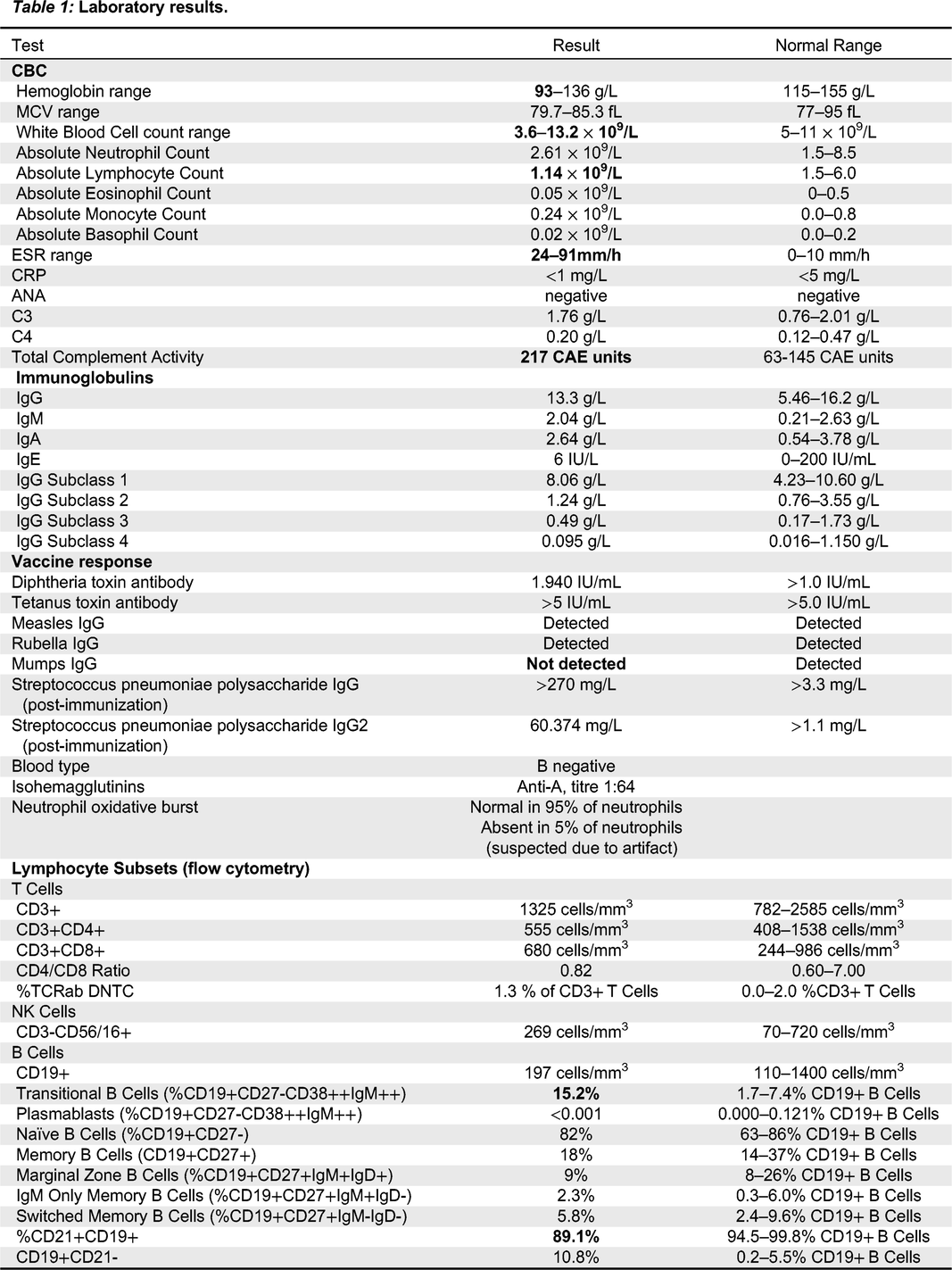
Our patient’s presentation of oro-genital mucosal ulcers, recurrent fevers, recurrent sinopulmonary infection and heterozygous 6q23.2-23.3 deletion encompassing TNFAIP3 was consistent with HA20 and she was initiated on colchicine.
Discussion: Our patient’s presentation is consistent with other reported cases of HA20 in the literature (Zhou et al. 2016; Aeschlimann et al. 2018; Franco-Jarava et al. 2018). In addition to her fevers and ulcers, she had developmental delay, behavioural concerns, short stature, hearing loss and recurrent sinopulmonary infection, though it unclear to what extent each of these features are related to underlying 6q deletion syndrome versus HA20.
In addition to TNFAIP3, other genes associated with known disease within the area of deletion include MYB, AHI1, PEX7, and IFNGR1, although there are a number of other genes in this region with unknown function. Defects in IFNGR1 can cause autosomal recessive recurrent mycobacterial infections. Although functional testing of STAT1 phosphorylation was not pursued in our patient, her deletion was heterozygous, and she had no features of increased susceptibility to mycobacterial infection.
HA20 is known to disrupt regulation of multiple immunological pathways involving inflammation and immunity (Dutrannoy et al. 2009). A20 is a potent inhibitor of the NF-kB signalling pathway, and restricts inflammation via its deubiquitinase activity (Dutrannoy et al. 2009). TNF binds to TNFR (TNF receptor), which leads to ubiquitination of RIP1. A20 deubiquitinates RIP1, preventing RIP1 from interacting with the NF-kB essential modulator (NEMO), stopping the proinflammatory response. A20 also adds polyubiquitin chains to another site on RIP1, which targets RIP1 for degradation, further limiting proinflammatory signalling (Dutrannoy et al. 2009).
A significant subset of patients with HA20 has been reported to suffer from recurrent infections (9%, or 8/89 patients in this cohort) (Chen et al. 2020), and several have been reported to have IgG subclass deficiency and absent polysaccharide vaccination response, which could explain the predisposition to sinopulmonary infection (nearly 50%, or 7/16 patients in this cohort) (Aeschlimann et al. 2018). Some have been treated with immune globulin replacement therapy. However, our patient demonstrated intact response to polysaccharide vaccination, and normal IgG subclasses.
Other immune abnormalities described in individuals with HA20 include low class-switched memory B cells, elevated “immune-exhausted” C21lowCD38low B cells, elevated transitional B cells, specific antibody deficiency, production of autoantibodies, inverted CD4:CD8 ratio, and expansion of Th17 cells (Hautala et al. 2020). Our patient had an increased proportion of transitional B cells and reduced proportion of CD21+CD19+ B cells, but no indication of decreased class-switched memory B cells, inverted CD4:CD8ratio, or increased immune exhausted B cells. Although Th17 cells have not yet been checked in our patient, interestingly, expansion of Th17 cells was not seen in the one other patient with the same 6q23.2q23.3 as our patient (Viel et al. 2018). At least one case in the literature describes a patient with HA20 and inflammasome-mediated lung disease responsive to anti-IL1 treatment, although our patient’s lung symptoms responded to antibiotics and thus have not been felt to be inflammatory in nature (Hautala et al. 2020).
Seven other cases of HA20 caused by chromosomal deletions have been previously reported (5 with large contiguous 6q deletions, of whom 1 had an identical deletion to our patient) (see Table 2). However, as has been noted previously, the reports in the literature for these patients have focused on their neurodevelopmental features rather than their immunologic phenotypes (Wu et al. 2021). Interestingly, recurrent sinopulmonary infections were reported in 4 of these cases, and infection was a presenting feature of at least one, as well as the present case. However, the other patient whose first symptom was recurrent infections did not have a reported immune work up. Detailed immunologic workup was only reported for 3 of the other published cases. The immune workup for our patient indicated an essentially intact humoral immune system, with the main immunologic findings being elevated markers of inflammation (ESR, CRP), increased proportion of transitional B cells and reduced proportion of CD21+CD19+ B cells.
Table 2:
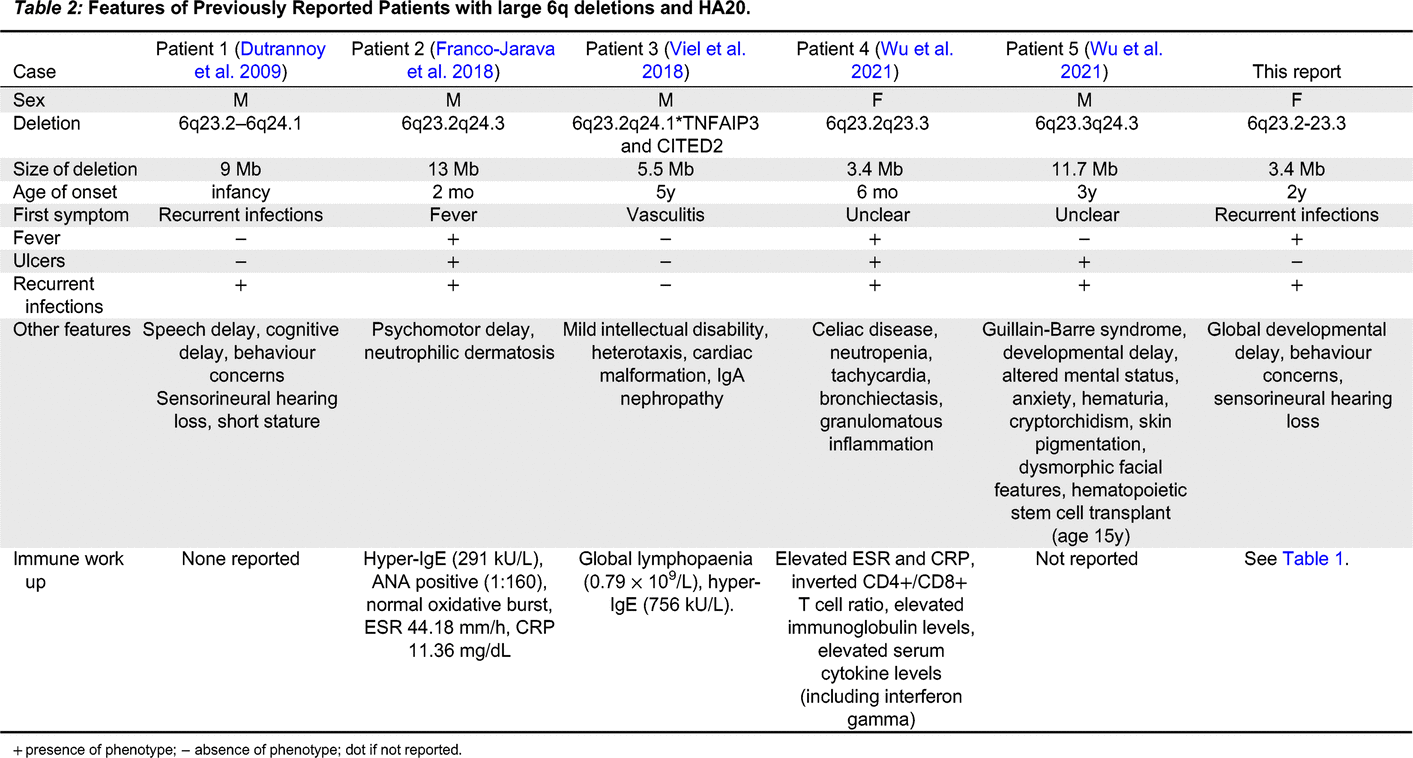
+ presence of phenotype; − absence of phenotype; dot if not reported.
Wu et al. noted that patients with HA20 due to contiguous gene deletions more frequently exhibit abdominal pain, lymphadenopathy, recurrent infections, short stature, failure to thrive, IUGR, speech delay, and/or intellectual disability than patients who have SNVs or indels in TNFAIP3 alone (Wu et al. 2021). Indeed, our patient demonstrated many of these features, including recurrent infection, short stature, failure to thrive, speech delay, and intellectual disability. Similar to the two HA20 patients recently reported by Wu et al. our patient also did not demonstrate arthritis. Additional investigations are pending to determine if our patient has gastrointestinal ulcerations, and to assess their Th17 cells.
Immune system dysfunction is known to be common amongst patients with chromosomal abnormalities, and some syndromes, such as Down and DiGeorge, are well known to have underlying immunodeficiency (Kusters et al. 2009; Davies 2013). In a retrospective, observational survey of 46 patients with chromosomal abnormalities (excluding DiGeorge and Down syndrome) and recurrent infections, 43 of 46 were reported to have recurrent ear-nose-throat infections. Antibody deficiency was the most common immunologic abnormality found, occurring in 33 of 46 patients, and 37 of 46 patients had associated developmental delay (Schatorjé et al. 2016). The apparent predisposition to recurrent infection in HA20, which appears to possibly be even greater in cases of HA20 caused by chromosomal deletions, further highlights the importance of immune system evaluation in patients with chromosomal abnormalities and apparent predisposition to infection or other evidence of immune dysregulation.
Conclusions: Haploinsufficiency A20 is an autoinflammatory syndrome where recurrent oto-sinopulmonary infection may be an early, persistent or even predominant feature of the disease. We presented a case of HA20 caused by a large contiguous chromosomal deletion, where diagnosis was made many years after initial microarray was performed, only after the clinical function of TNFAIP3 was elucidated.
Our case highlights the importance of immune evaluation in individuals with chromosomal deletions and developmental delay, as well as the value of revisiting previously performed genetic testing results in patients with unexplained clinical features, as genetic and phenotypic knowledge is constantly expanding.
REFERENCES
Aeschlimann, F.A., Batu, E.D., Canna, S.W., Go, E., Gül, A., Hoffmann, P., Leavis, H.L., Ozen, S., Schwartz, D.M., Stone, D.L., van Royen-Kerkof, A., Kastner, D.L., Aksentijevich, I., and Laxer, R.M. 2018. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Annal. Rheumatic Dis. 77(5): 728–735. doi: 10.1136/annrheumdis-2017-212403.
Catrysse, L., Vereecke, L., Beyaert, R., and van Loo, G. 2014. A20 in inflammation and autoimmunity. Tren. Immunol. 35(1): 22–31. doi: 10.1016/j.it.2013.10.005.
Chen, Y., Ye, Z., Chen, L., Ye, Z., Chen, L., Qin, T., Seidler, U., Tian, D., and Xiao1, F. 2020. Association of Clinical Phenotypes in Haploinsufficiency A20 (HA20) With Disrupted Domains of A20. Front Immunol. 11: 574992. doi: 10.3389/fimmu.2020.574992.
Davies, E.G. 2013. Immunodeficiency in DiGeorge Syndrome and Options for Treating Cases with Complete Athymia. Front Immunol. 4: 322. doi: 10.3389/fimmu.2013.00322.
Dutrannoy, V., Klopocki, E., Wei, R., Bommer, C., Mundlos, S., Graul-Neumann, L.M., and Trimborn, M. 2009. De novo 9 Mb deletion of 6q23.2q24.1 disrupting the gene EYA4 in a patient with sensorineural hearing loss, cardiac malformation, and mental retardation. Europ. J. Medical Gene. 52(6): 450–453. doi: 10.1016/j.ejmg.2009.06.004.
Franco-Jarava, C., Wang, H., Martin-Nalda, A., Alvarez, de S.D., García-Prat, M., Bodet, D., García-Patos, V., Plaja, A., Rudilla, F., Rodriguez-Sureda, V., García-Latorre, L., Aksentijevich, I., Colobran, R., and Soler-Palacín, P. 2018. TNFAIP3 haploinsufficiency is the cause of autoinflammatory manifestations in a patient with a deletion of 13Mb on chromosome 6. Clin. Immunol. 191: 44–51. doi: 10.1016/j.clim.2018.03.009.
Hautala, T., Vähäsalo, P., Kuismin, O., Keskitalo, S., Rajamäki, K., Väänänen, A., Simojoki, M., Säily, M., Pelkonen, I., Tokola, H., Mäkinen, M., Kaarteenaho, R., Jartti, A., Hautala, N., Kantola, S., Jackson, P., Glumoff, V., Saarela, J., Varjosalo, M., Kulund, K.K., and Seppänen, M.R.J. 2020. A Family With A20 Haploinsufficiency Presenting With Novel Clinical Manifestations and Challenges for Treatment. JCR: J. Clin. Rheumatol. Publish Ahead of Print. doi: 10.1097/RHU.0000000000001268.
Kusters, M.A., Verstegen, R.H.J., Gemen, E.F.A., andVries, E.D. 2009. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin. Exper. Immunol. 156(2): 189–193. doi: 10.1111/j.1365-2249.2009.03890.x.
Schatorjé, E., van der Flier, M., Seppänen, M., Browning, M., Morsheimer, M., Henriet, S., Neves, J.F., Vinh, D.C., Alsina, L., Grumach, A., Soler-Palacin, P., Boyce, T., Celmeli, F., Goudouris, E., Hayman, G., Herriot, R., Förster-Waldl, E., Seidel, M., Simons, A., and de Vries, E. 2016. Primary immunodeficiency associated with chromosomal aberration – an ESID survey. Orphanet J. Rare Dis. 11: 110. doi: 10.1186/s13023-016-0492-1.
Viel, S., Cheyssac, E., Pescarmona, R., Besson, L., Till, M., Viremouneix, L., Touitou, I., Sarrabay, G., Walzer, T., and Belot, A. 2018. Large deletion in 6q associated to A20 haploinsufficiency and thoracoabdominal heterotaxy. Ann. Rheum. Dis. 77(11): 1697–1698. doi: 10.1136/annrheumdis-2018-213300.
Wu, C.W., Sasa, G., Salih, A., Nicholas, S., Vogel, T.P., Cahill, G., Kuehn, H.S., Rosenzweig, S.D., Zhou, Q., Chinn, I.K., and Yuan, B. 2021. Complicated Diagnosis and Treatment of HA20 due to Contiguous Gene Deletions involving 6q23.3. J. Clin. Immunol. 41(6): 1420–1423. doi: 10.1007/s10875-021-01048-w.
Zhou, Q., Wang, H., Schwartz, D.M., Stoffels, M., Park, Y.H., Zhang, Y., Yang, D., Demirkaya, E., Takeuchi, M., Tsai, W.L., Lyons, J.J., Yu, X., Ouyang, C., Chen, C., Chin, D.T., Zaal, K., Chandrasekharappa, S.C., Hanson, E.P., Yu, Z., Mullikin, J.C., Hasni, S.A., Wertz, I.E., Ombrello, A.K., Stone, D.L., Hoffmann, P., Jones, A., Barham, B.K., Leavis, H.L., Royen-Kerkof, A.V., Sibley, C., Batu, E.D., Gül, A., Siegel, R.M., Boehm, M., Milner, J.D., Ozen, S., Gadina, M., Chae, J., Laxer, R.M., Kastner, D.L., and Aksentijevich, I. 2016. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early onset autoinflammatory syndrome. Nat. Genet. 48(1): 67–73. doi: 10.1038/ng.3459.
Immunohistopathology evaluation of an X-MAID patient with a novel mutation in MSN
Laura Abrego Fuentes1, Amarilla B. Mandola2, Bo Ngan3, Chaim M. Roifman1,4
1Division of Immunology & Allergy, Department of Pediatrics, Hospital for Sick Children and University of Toronto, Toronto, ON
2Pediatric Dept A, Soroka University Medical Center, Ben-Gurion University of the Negev, Beer-Sheva, Israel
3Department of Paediatric Laboratory Medicine, Hospital for Sick Children, Toronto, ON
4Canadian Centre for Primary Immunodeficiency, Hospital for Sick Children, Toronto, ON
Background: The cell cytoskeleton is finely regulated by the ezrin-radixin-moesin (ERM) family of proteins, which form structural links between transmembrane proteins and the underlying actin cytoskeleton (Fehon et al. 2010). They are essential for normal membrane organization and function, including maintenance of cell shape, microvilli formation, pseudopod/uropod and immune synapse formation, phagocytosis and apoptosis, as well as regulation of signal transduction pathways (Niggli and Rossy 2008). Phosphorylation and subsequent activation of ERM proteins enables interactions with partners critically involved in shape regulation, such as actin filaments, transmembrane proteins, and scaffolding proteins.
Moesin is encoded by the MSN gene located on the X-chromosome. It is ubiquitously expressed in lungs, spleen, kidney, endothelial cells of vessels, and is the predominant ERM protein in lymphocytes and neutrophils (Liu et al. 2015). Deficiency or dysregulation of moesin has been shown to disrupt the development of lymphoid cells and neutrophils (Lagresle-Peyrou et al. 2016; Liu et al. 2015)
The recently described X-linked moesin-associated immunodeficiency (X-MAID), part of a group of immunodeficiencies caused by defects in cytoskeletal regulation, is characterized by severe leukopenia affecting T cells, B cells, and neutrophils (Lagresle-Peyrou et al. 2016). To date, the clinical picture of patients with X-MAID varies from those requiring minimal intervention, through to immunoglobulin replacement therapy and antibiotic prophylaxis, and allogeneic transplantation (Lagresle-Peyrou et al. 2016; Henrickson et al. 2019; Bradshaw et al. 2018).
Here, we describe the presentation, immune-workup, and histopathology findings of a young male patient with X-MAID and multi-organ involvement, whose severe pulmonary vein stenosis necessitated a double lung transplant.
Methods: A thorough review of the patient’s chart was performed. The patient was enrolled in the Primary Immunodeficiency Registry and Tissue Bank (REB protocol number 1000005598) for additional research-based evaluations.
Results:
Case presentation: Our patient is currently an 8-year-old male from a non-consanguineous Caribbean family who presented to our Immunology Service at age 3 with a history of recurrent infections, including recurrent respiratory tract, oral thrush and 3 major bacterial infections: Streptococcal bacteremia at age 2 years, group A Strep. pharyngitis at age 3 years, and periorbital cellulitis at age 3.5 years, all requiring prolonged admission and intravenous antibiotic therapy. Additionally, he had high myopia, hypotonia, and motor developmental delay.
His immune evaluation revealed low CD3+ T cells, including both CD4+ and CD8+, and normal numbers of CD19+ and NK cells (Table 1). Other immune parameters were normal, including lymphocyte responses to phytohemagglutinin, as well as albumin and immunoglobulin levels, reactive specific antibody titers, NOBI, CH50, chromosomal microarray and chromosomal breakage assay.
At age 4 years he was admitted for severe pulmonary vein stenosis, leading to pulmonary hypertension, and subsequently underwent a double lung transplant. Presently, he continues to suffer from multiple infections, including recurrent pneumoniae (Staphlococcus, Pseudomonas, Hemophilus, Entero, Rhinovirus, MAC), and has been unable to clear Norovirus from his GI tract. Further, he has developed immune-mediated bicytopenia, alopecia areata, as well as chronic kidney injury post-transplant.
Genetic findings: Clinical trio whole exome sequencing revealed a novel hemizygous mutation in the MSN gene, encoding moesin. The variant, located in exon 4, introduces a premature stop codon and is predicted to cause loss of function—either through protein truncation or nonsense-mediated decay. Western blot analysis confirmed reduced expression of moesin in the patient’s peripheral blood lymphocytes.
Histopathology analysis: Histologic evaluation of the lung tissue prior to transplantation and detailed immune-histological evaluation of the thymus identified profound abnormalities in alveoli formation as well as marked signs of lymphoid underdevelopment, in keeping with a primary immunodeficiency.
Discussion: Moesin deficiency has only recently been described. Our detailed histopathology evaluation of the thymus and lungs represent the first documented for a patient with X-MAID who suffered severe pulmonary vein stenosis, necessitating a double lung transplant. Patients with moesin deficiency may present during infancy or childhood with a severe form of the disease, including combined immunodeficiency with lymphopenia and neutropenia, while adults may have a milder clinical picture (Lagresle-Peyrou et al. 2016). Other manifestations, including hypogammaglobulinemia and poor vaccine responses, have been reported. With the recent introduction of SCID newborn screening, X-MAID has also been identified via low levels of T cell receptor excision circles, demonstrating the importance of this screening tool (Delmonte et al. 2017; Dvorak et al. 2019).
Cumulative findings suggest a genotype-phenotype correlation among patients with X-MAID. Our patient, with a novel mutation in MSN, adds to the known spectrum of disease and highlights the non-redundant functions of moesin, particularly in the lung and immune organs.
Table 1:
REFERENCES
Bradshaw, G., Lualhati, R.R., Albury, C.L., Maksemous, N., Roos-Araujo, D., Smith, R.A., Benton, M.C., Eccles, D.A., Lea, R.A., Sutherland, H.G., Haupt, L.M., and Griffiths, L.R. 2018. Exome Sequencing Diagnoses X-Linked Moesin-Associated Immunodeficiency in a Primary Immunodeficiency Case. Front Immunol. 9: 420.
Delmonte, O.M., Biggs, C.M., Hayward, A., Comeau, A.M., Kuehn, H.S., Rosenzweig, S.D., and Notarangelo, L.D. 2017. First Case of X-Linked Moesin Deficiency Identified After Newborn Screening for SCID. J. Clin. Immunol. 37: 336–338.
Dvorak, C.C., Haddad, E., Buckley, R.H., Cowan, M.J., Logan, B., Griffith, L.M., Kohn, D.B., Pai, S.Y., Notarangelo, L., Shearer, W., Prockop, S., Kapoor, N., Heimall, J., Chaudhury, S., Shyr, D., Chandra, S., Cuvelier, G., Moore, T., Shenoy, S., Goldman, F., Smith, A.R., Sunkersett, G., Vander Lugt, M., Caywood, E., Quigg, T., Torgerson, T., Chandrakasan, S., Craddock, J., Davila Saldana, B.J., Gillio, A., Shereck, E., Aquino, V., Desantes, K., Knutsen, A., Thakar, M., Yu, L., and Puck, J.M. 2019. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J. Allergy. Clin. Immunol. 143: 405–407.
Fehon, R.G., Mcclatchey, A.I. and Bretscher, A. 2010. Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell. Biol. 11: 276–287.
Henrickson, S.E., Andre-Schmutz, I., Lagresle-Peyrou, C., Deardorff, M.A., Jyonouchi, H., Neven, B., Bunin, N., and Heimall, J.R. 2019. Hematopoietic Stem Cell Transplant for the Treatment of X-MAID. Front. Pediatr. 7: 170.
Lagresle-Peyrou, C., Luce, S., Ouchani, F., Soheili, T.S., Sadek, H., Chouteau, M., Durand, A., Pic, I., Majewski, J., Brouzes, C., Lambert, N., Bohineust, A., Verhoeyen, E., Cosset, F.L., Magerus-Chatinet, A., Rieux-Laucat, F., Gandemer, V., Monnier, D., Heijmans, C., van Gijn, M., Dalm, V.A., Mahlaoui, N., Stephan, J.L., Picard, C., Durandy, A., Kracker, S., Hivroz, C., Jabado, N., de Saint Basile, G., Fischer, A., Cavazzana, M., and Andre-Schmutz, I. 2016. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J. Allergy Clin. Immunol. 138: 1681–1689 e8.
Liu, X., Yang, T., Suzuki, K., Tsukita, S., Ishii, M., Zhou, S., Wang, G., Cao, L., Qian, F., Taylor, S., Oh, M.-J., Levitan, I., Ye, R.D., Carnegie, G.K., Zhao, Y., Malik, A.B., and Xu, J. 2015. Moesin and myosin phosphatase confine neutrophil orientation in a chemotactic gradient. J. Exp. Med. 212: 267–280.
Niggli, V., and Rossy, J. 2008. Ezrin/radixin/moesin: Versatile controllers of signaling molecules and of the cortical cytoskeleton. Inter. J. Biochemis. Cell Biol. 40: 344–349.
A case of Trichohepatoenteric Syndrome due to biallelic damaging variants in TTC37A
Meriem Latrous1, Elliot James1, Catherine M. Biggs1,2, Stuart E. Turvey1,2, Kyla J. Hildebrand1,2
1Division of Allergy and Immunology, Department of Paediatrics, BC Children’s Hospital, The University of British Columbia, Vancouver, BC, Canada
2British Columbia Children’s Hospital Research Institute; Vancouver, BC, Canada
Case:
A female infant was born to a 37-year-old G2P1 mother via urgent c-section at 34 plus 3 weeks’ gestational age for abnormal doppler ultrasound. The pregnancy was complicated by severe symmetric intrauterine growth restriction (IUGR), worsening oligohydramnios, suspected small kidneys, cardiomegaly, and an umbilical vein varix. There was no history of maternal illness and serologies were protective. Initial Serum Integrated Prenatal Screening (SIPS) was abnormal and thought to be representative of placental dysfunction. An amniocentesis was performed demonstrating a normal female karyotype and chromosomal microarray, and a negative TORCH screen. After delivery, positive pressure ventilation was required for poor respiratory effort after which she was transitioned to continuous positive airway pressure therapy (CPAP). Apgars at birth were 6, 7, and 8 at one, five and ten minutes respectively. All growth parameters were below the 3rd percentile, with a very low birthweight of 1050g and head circumference of 27 cm. The neonate was transferred to the Neonatal Intensive Care Unit (NICU) for ongoing support and was eventually weaned off CPAP. A postnatal echocardiogram identified multiple small apical muscular ventricular septal defects (VSD) with no other abnormalities.
The post-natal course in the NICU was complicated, and Immunology was consulted on day of life 67 (corrected age, 3 weeks) for possible underlying inborn error of immunity in the setting of persistent unexplained lymphopenia, failure to thrive, and chronic non-bloody diarrhea requiring total parental nutrition (TPN). Parents were healthy, non-consanguineous and of Caucasian backgrounds of Swedish, Irish, and Scottish (maternal), and Bulgarian (paternal) descent. A two-year-old male sibling was born at term and was healthy. Extended family history was negative for inborn errors of immunity, autoimmune conditions, recurrent miscarriages, or unexplained deaths in children. A paternal grandfather died of leukemia at age 52 years.
Physical examination revealed growth parameters well below the first percentile for head circumference (33 cm), length (43 cm), and weight (1839 g). Facial features were in keeping with a premature baby with IUGR. Mild hypertelorism was noted however interpupillary distance was not measured. There were no other dysmorphic features. Examination of the oropharynx revealed no evidence of oral thrush. There were no palpable cervical, inguinal, or axillary lymph nodes. Respiratory, cardiovascular, and abdominal examinations were normal. The nails and skin were within normal limits; however, the hair was very fine, sparse and with a “frizzy” texture.
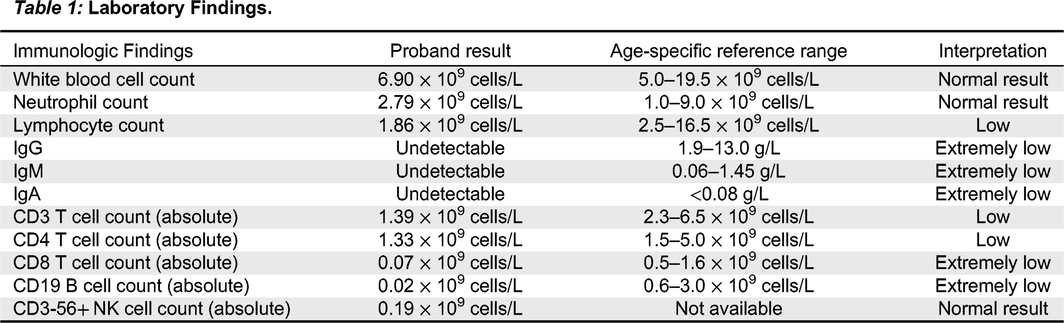
Lymphocyte counts since birth had been low at less than 2 × 109 cells/L (normal range 2.5–16.5 × 109 cells/L). A summary of laboratory findings at the time of assessment can be found in Table 1. A T-cell memory panel was unremarkable for both CD4 and CD8, however this was unreliable given the very low absolute number of CD8 T-cells. Dihydrorhodamine Flow Cytometric Assay was normal. T-cell receptor excision circle (TREC) assay was sent from the patient’s newborn screen to Ontario and was initially low. A repeat assay was just above the lower limit of the cut-off value at 88 (normal range >75).
Table 1:
An infectious disease work up included negative results for presence of viruses and bacteria in cultures (blood, urine, cerebrospinal fluid). A nasopharyngeal swab was positive for enterovirus and rhinovirus that persisted on several sample collections over time. Newborn screening was positive for Hypermethioninemia, and although this was initially thought to be falsely positive due to Total Parenteral Nutrition (TPN) administration, a repeat screen performed 3 weeks later remained positive. Confirmatory testing with plasma amino acid testing revealed a methionine level of 374 umol/L (normal range 19–49 umol/L). Review of chest x-rays from birth were suggestive of an absent thymus. An abdominal ultrasound revealed multiple hypoechoic nodules concerning for a possible fungal infection.
Given the history of prematurity, severe IUGR, microcephaly, and postnatal course complicated by failure to thrive, chronic diarrhea, persistent unexplained lymphopenia, viral infection, undetectable IgG, IgA, IgM, as well as very low T-cells and B-cells, there was significant concern for a diagnosis of T-B-NK+severe combined immunodeficiency (SCID). The liver nodules were concerning for a possible fungal infection and treatment with Amphotericin B was initiated, along with intravenous immunoglobulin replacement therapy and trimethoprim/sulfamethoxazole for PJP prophylaxis.
Rapid Whole Exome Sequencing as a trio revealed two likely pathogenic variants in the TCC37 gene: c.2578-7_2578-3del intronic variant (maternally inherited and predicted to result in loss of the canonical acceptor splice site) and c.1325+1G>C splice site variant (de novo and disrupting the consensus GT splice donor site). The genetic results along with the clinical presentation of intractable congenital diarrhea, combined immunodeficiency, IUGR/failure to thrive, and hypermethioninemia established the diagnosis of Trichohepatoenteric syndrome (THES). Dermatology was subsequently consulted and identified trichorrhexis nodosa upon microscopic review of a hair sample (Figure 1).
Figure 1:

Fungal cultures of liver biopsy samples were negative. Direct Internal Transcribed Spacer (ITS) Polymerase chain reaction (PCR) analysis of the same samples identified presence of Candida species; however, specimen contamination could not be ruled out. The liver nodules worsened on serial ultrasounds while receiving antifungal therapy, suggesting intrinsic liver disease and cirrhosis. Over the next few months, the infant developed worsening cholestasis, liver dysfunction, and TPN dependence. She subsequently died of fulminant Escherichia coli (E. coli) sepsis at 5 months of life (corrected age 13 weeks).
Discussion: Trichohepatoenteric syndrome (THES), also known as Phenotypic Diarrhea of Infancy, is a rare disorder inherited in an autosomal recessive pattern. It has been described in approximately 52 patients to date and was first reported in 1994. Most cases are due to variants in the TTC37 gene, but variants in the Ski2 Like RNA Helicase gene (SKIV2L) have also been found to be disease-causing (Fabre et al. 2018). Protein expression is found intracellularly, in a variety of organs including the placenta, kidneys, brain, gastrointestinal tract, liver, lung, lymph nodes and vascular tissue. The oldest patients reported with this condition are in their late 20's, with few individuals in late adolescence. Of the known reported cases, there have been 21 deaths with infections implicated in at least 7 patients (Fabre et al. 2018).
THES is a multisystem disorder often presenting with IUGR, growth failure, woolly, or brittle hair with trichorrhexis nodosa, and severe infantile diarrhea that is typically lifelong and with onset at usually less than 6 months of age. In some cases, this may present similarly to very early onset-IBD. Facial dysmorphisms, including hypertelorism, a broad nasal root, and prominent cheeks/forehead may become more evident with age. Liver disease may also occur with elevated aminotransferases, hepatomegaly, and risk of fibrosis/cirrhosis. Hypermethioninemia has been described. In some cases, progression to hemochromatosis has occurred. Developmental or cognitive delay (typically mild) occurs in approximately 50% of affected patients. Dermatologic manifestations include cafe-au-lait macules and dyschromic spots. Rare manifestations include congenital heart disease, platelet abnormalities (typically without bleeding diathesis), and Hemophagocytic Lymphohistiocytosis (Fabre et al. 2018).
The immunological defect in THES is significant and can affect B and T lymphocytes and NK cells. In our patient, enterovirus, and rhinovirus, both single-stranded RNA viruses, were identified in nasopharyngeal samples. The infant also eventually developed fulminant E. coli sepsis. Laboratory findings were in keeping with previous observations in this disorder, including absent immunoglobulins, low T lymphocytes, and extremely low CD19 B-cells.
Vely et al. describe three immune defects in THES including low switched memory B, impaired IFN-γ production by T and NK cells associated with reduced degranulation of NK cells, and abnormal T cell proliferation (Vély et al. 2018). Recurrent infections with Epstein-Barr Virus, Respiratory syncytial virus, and bacteria have been described. The exact mechanism of susceptibility to these pathogens is unclear, however TTC37 and SKIV2L seem to have a role in RNA surveillance and degradation via the RNA exosome, an evolutionarily conserved ribonuclease complex that is critical for both processing and degradation of a variety of RNAs (Fabre et al. 2011, 2012; Hartley et al. 2010; Morton et al. 2017). TTC37 has been reported in the literature as the ortholog of yeast SKI3, encoding an important component of the Ski complex, which is required for exosome-mediated RNA surveillance (Fabre et al. 2012; Brown et al. 2000). Though THES is associated with at least two different genes, its clinical manifestations are indistinguishable which suggests that a defect in Ski-complex function or structure underlies the main clinical features (Fabre et al. 2012).
While this may be life-limiting in some, there are also patients described with long-term survival, and it is not possible to specifically predict the outcome or course for any given individual. Mainstays of treatment include TPN and close nutritional monitoring. Oral feeding (e.g., elemental, or amino acid-based formulas) is encouraged as tolerated, and in some cases, weaning off TPN has been described (in the literature, around age 15 months–3 years). Hematopoietic Stem Cell Transplant (HSCT) has been performed in two patients to date: one of which died of infection and the other who had no improvement in symptoms (Fabre et al. 2018).
In our patient, autopsy confirmed cirrhosis with interval progression of hepatic damage from previous biopsies. Neuropathological examination demonstrated a small brain. Firm white liver nodules were present with features of infantile hemangioendothelioma, and the amount of germ cells present in the ovaries appeared markedly reduced compared to age-matched controls, which are features that have not been previously described in THES. An unexpected finding was a remarkably short small bowel, approximately 25% of the expected length. This has also not been previously described in THES and may have contributed to persistent diarrhea.
Conclusion: We present a case of an infant with clinical features suspicious for inborn error of immunity, diagnosed with Trichohepatoenteric Syndrome on Whole Exome Sequencing. Althouh a very rare condition, it is important to consider this diagnosis in patients presenting with combined immunodeficiency, growth failure, diarrhea, and liver disease. Though the clinical phenotype may resemble SCID, the management of this disorder is significantly different, and a molecular diagnosis will facilitate prognosis counselling for families. Our patient had a significantly short bowel, infantile hemangioendotheliomas, and reduced presence of ovarian germ cells, which have not previously been observed in this syndrome and may represent additional phenotypic findings in this disease.
REFERENCES
Brown, J.T., Bai, X., and Johnson, A.W. 2000. The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA. 6: 449–457.
Fabre, A., Martinez-Vinson C., Roquelaure B., Missirian, C., André, N., Breton, A., Lachaux, A., Odul, E., Colomb, V., Lemale, J., Cézard, J-P., Goulet, O., Sarles, J., Levy, N., and Badens, C. 2011. Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome. Hum. Mutat. 32(3): 277–281.
Fabre, A., Charroux, B., Martinez-Vinson, C., Roquelaure, B., Odul, E., Sayar, E., Smith, H., Colomb, V., Andre, N., Hugot, J-P., Goulet, O., Lacoste, C., Sarles, J., Royet, J., Levy, N., and Badens, C. 2012. SKIV2L Mutations Cause Syndromic Diarrhea, or Trichohepatoenteric Syndrome. Am. J. Hum. Gene. 90: 689–692.
Fabre, A., Bourgeois, P., Chaix, C., Bertaux, K., Goulet, O., and Badens, C. 2018. Trichohepatoenteric Syndrome. In GeneReviews®. Edited by M.P. Adam, H.H. Ardinger, R.A. Pagon, S.E. Wallace, L.J.H. Bean, G. Mirzaa, and A. Amemiya. Seattle (WA): University of Washington, Seattle.
Hartley, J.L., Zachos, N.C., Dawood, B., Donowitz, M., Forman, J., Pollitt, R.J., Morgan, N.V., Tee, L., Gissen, P., Kahr, W.H.A., Knisely, A.S., Watson, S., Chitayat, D., Booth, I.W., Protheroe, S., Murphy, S., de Vries, E., Kelly, D.A., and Maher, E.R. 2010. Mutations in TTC37 cause Trichohepatoenteric Syndrome (phenotypic diarrhea of infancy). Gastroenterol. 138(7): 2388–2398.e23982.
Morton, D.J., Kuiper, E.G., Jones, S.K., Leung S.W., Corbett, A.H., and Fasken, M.B. 2017. The RNA exosome and RNA exosome-linked disease. RNA. 24: 127–142.
Vély, F., Barlogis, V., Marinier, E., Coste, M.E., Dubern, B., Dugelay, E., Lemale, J., Martinez-Vinson, C., Peretti, N., Perry, A., Bourgeois, P., Badens, C., Goulet, O., Hugot, J.P., Farnarier, C., and Fabre, A. 2018. Combined Immunodeficiency in Patients with Trichohepatoenteric Syndrome. Front Immunol. 9: 1036.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 4 • December 2021
Pages: 105 - 127
History
Version of record online: 10 December 2021
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
2021. Abstracts from the Immunodeficiency Canada—9th SCID Symposium, 28 October 2021. LymphoSign Journal.
8(4): 105-127. https://doi.org/10.14785/lymphosign-2021-0028
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
