A novel splice site variant in FOXN1 in a patient with abnormal newborn screening for severe combined immunodeficiency and congenital lymphopenia
Abstract
Background: The Forkhead box protein N1 (FOXN1) is a key regulator of thymic epithelial development, and its complete deficiency leads to a nude-severe combined immunodeficiency (SCID) phenotype. More recently, heterozygous mutations in FOXN1 have been linked with a syndrome of congenital lymphopenia and a wide clinical spectrum, with most cases being caused by missense mutations.
Aim: To broaden the genotypic and phenotypic spectrum of heterozygous FOXN1 deficiency.
Methods: Case report of a patient with FOXN1 haploinsufficiency due to a novel splice-site mutation.
Results: Our patient was identified at 3 weeks of life given an abnormal newborn screen (NBS) for SCID, and was found to have congenital lymphopenia preferentially affecting CD8+ T-cells. Her cellular and humoral function were both excellent, and she has remained entirely asymptomatic and thriving for the first 3 years of her life. The patient was found on whole exome sequencing to carry a heterozygous splice-site mutation in the FOXN1 gene, affecting the Forkhead domain. The mutation was also identified in her asymptomatic mother.
Conclusion: Heterozygous FOXN1 mutations are an increasingly-recognized cause of congenital lymphopenia. Our experience suggests most patients remain clinically well, with main manifestation including T-lymphopenia, mostly affecting CD8+ cells. Identification of the same variant in an asymptomatic parent suggests age-dependent improvement in T-cell counts and an overall benign course, while provides impetus for diligent conservative management with regular follow-up.
Statement of novelty: Heterozygous FOXN1 deficiency is a relatively new entity, attributed in most cases to missense mutations in FOXN1. To further expand the knowledge basis regarding this emerging disorder, as well as its genotypic repertoire, we herein report a case of heterozygous FOXN1 deficiency caused by a splice site mutation.
Introduction
The Forkhead box (FOX) superfamily comprises of transcription factors with key roles in tissue development and homeostasis (Lam et al. 2013). Within the FOX family, FOX protein N1 (FOXN1) is a key regulator of thymic epithelium and skin development. FOXN1 dictates gene expression involved in thymic epithelial cell (TEC) differentiation, lymphoid progenitor migration from the bone marrow, antigen processing and thymocyte selection (Nowell et al. 2011; Romano et al. 2013; Žuklys et al. 2016). Complete FOXN1 deficiency was first identified in mice displaying the nude/severe combined immunodeficiency (SCID) phenotype, consisting of congenital alopecia totalis, nail dystrophy and thymic aplasia (Flanagan 1966). An equivalent phenotype was later described in humans harbouring biallelic loss-of-function (LOF) mutations in the FOXN1 gene (Pignata et al. 1996; Frank et al. 1999). In contrast to the well-established phenotype seen with complete FOXN1 LOF, the role of heterozygous FOXN1 mutations in human immunity has been slower to emerge. In a recent case-series, Bosticardo et al. (2019) described for the first time a cohort of children with heterozygous FOXN1 mutations, who were found to have low T-cell receptor excision circles (TREC) levels and (or) lymphopenia.
Functional and clinical presentation
Clinical case
An asymptomatic female newborn was referred to our Immunology clinic at the age of 3 weeks by the Ontario newborn screening (NBS) program. The reason for referral was a finding of low T-cell receptor excision circles (TREC) levels from a dried blood spot, representing an abnormal screening test for severe combined immunodeficiency (SCID). On review of history, the girl was the product of spontaneous pregnancy with adequate prenatal follow-up and no known pregnancy complications. She was born at 39 + 2 weeks via spontaneous vaginal delivery with no resuscitation required, and had an uneventful neonatal course. Family history identified non-consanguineous parents of mixed English, Dutch, and African-American ancestry. Father had required a tonsillectomy and adenoidectomy as a child for recurrent tonsillitis, and had a number of acute otitis media infections until the age of 4 years, for which he did not require any surgical intervention. Mother was healthy with no history of recurrent infections. Two maternal half siblings were healthy as well. There was no extended family history of immunodeficiency, autoimmunity, inflammation, or early-onset malignancy. When seen at our clinic at the age of 3 weeks, the girl had been growing and feeding well and had no notable concerns for infection, autoimmunity, or inflammation. Her physical exam was within normal limits, including normal growth parameters, presence of normal lymph nodes and tonsillar tissue, no adenopathy, hepatosplenomegaly, or skin rashes; in particular, she has not demonstrated any alopecia or dermatitis.
Investigations
TREC levels were low, both on initial testing (48 copies/3 μL; cutoff: ≥75), and on repeat testing 1 month later (41.3 copies/3 μL). TREC measurement has not been repeated since. Adenosine deaminase and purine nucleoside phosphorylase metabolite profiles were normal. Complete blood count and differential were normal, including a normal absolute lymphocyte count of 2.55 × 109/L (normal: 2–17 × 109/L). Lymphocyte immunophenotyping, however was abnormal, with low CD3+CD4+ (837 cells/μL; normal: 1700–5300 cells/μL) and CD3+CD8+ (178 cells/μL; normal: 400–1700 cells/μL) counts. CD19+ cells were normal (903 cells/μL; normal: 600–1900 cells/μL), as were CD16+CD56+ cells (438 cells/μL; normal: 186–724 cells/μL). Total immunoglobulin levels, PHA stimulation index, T-cell receptor Vβ repertoire, and analysis of naïve/memory T-cells (CD45RA/RO) were all within normal limits. On subsequent evaluations over the following 3 years, the she continued to have evidence of T-cell lymphopenia preferentially affecting CD3+CD8+, though this has remained in the moderate range. Her B and NK cells remain unaffected, and her functional assessment continues to be excellent for both the humoral and cellular arms. Following administration of killed immunizations, normal specific antibody titres were noted to both diphtheria and tetanus toxoid.
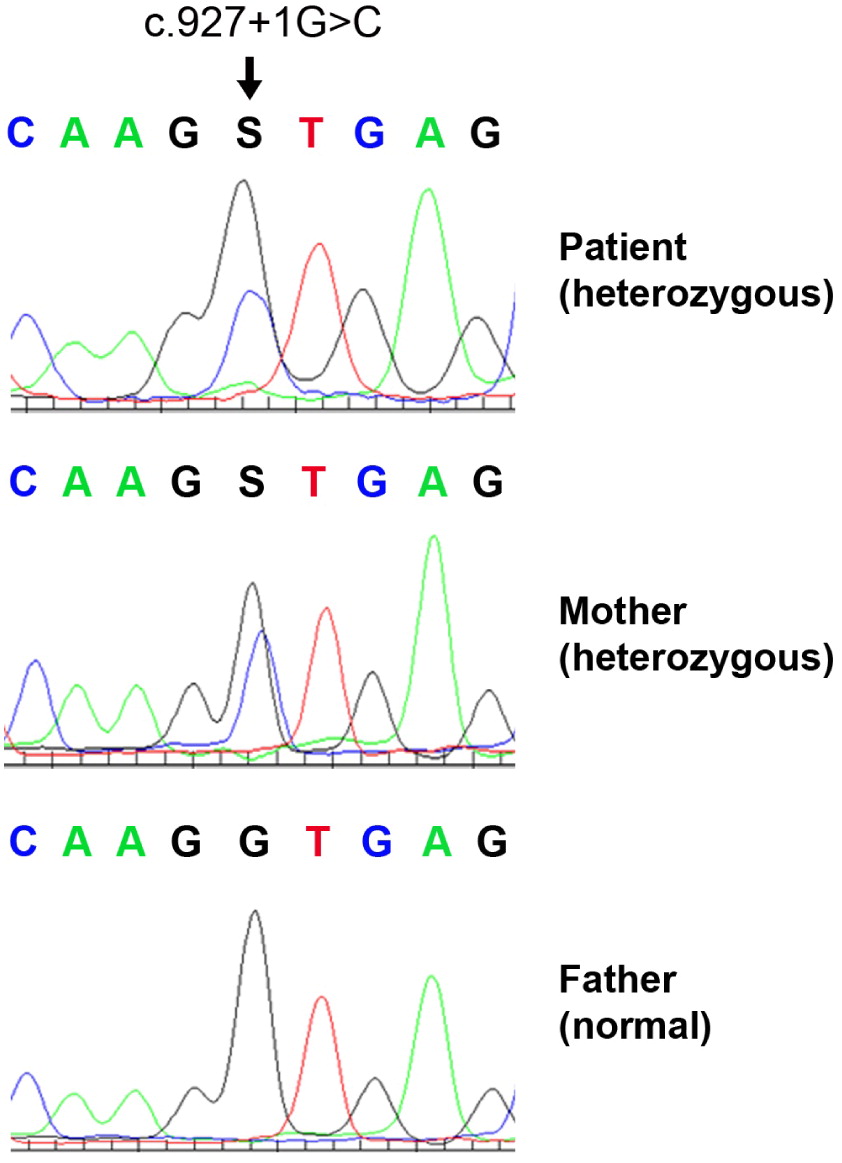
Genetic analysis began with normal karyotype and fluorescent in-situ hybridization for 22q11.2. Further analysis involved a primary immunodeficiency panel, which did not reveal the cause of the patient’s presentation. Whole exome sequencing was finally performed, revealing a novel heterozygous intronic variant of unknown significance in the FOXN1 gene: NM_003593 c.927+1G>C. The variant has not been reported in large population databases, and is predicted to eliminate an obligatory donor splice site in the Forkhead domain resulting in a deleterious effect. The variant was also detected in the girl’s mother, and not in her father (Figure 1). On further testing, mother demonstrated a normal complete blood count and differential.
Figure 1:
Outcome
Over the following 3 years, the patient has continued to be well with no recurrent or unusual infections, growth retardation, or immune dysregulation. She continues to have T-lymphopenia preferentially affecting CD8+ T-cells, although this has remained in the moderate range, with excellent T-cell stimulation responses. She is soon due to begin her live viral immunization series.
Discussion
The implementation of NBS in Ontario, as well as in various provinces across Canada, has led to an increased detection rate of patients with non-SCID lymphopenia who display low TREC levels shortly after birth. In this regard, heterozygous FOXN1 mutations have been recently identified as a genetic etiology in a subset of children with abnormal NBS and (or) lymphopenia. The current report contributes to the expanding spectrum of heterozygous FOXN1 deficiency, both genotypically and phenotypically. Clinically, our patient has always been well, and would likely have not presented to Immunology had it not been for her abnormal NBS result. She has had persistent moderate CD8+ lymphopenia and mild and improving CD4+ lymphopenia, with normal T-cell function and no humoral abnormalities. The patient was found to carry a heterozygous splice-site mutation in FOXN1, likely resulting in a loss of function. Interestingly, her mother who is clinically well and had a normal absolute lymphocyte count as an adult, was found to carry the same mutation. This may be attributed to the age-dependent function of FOXN1, which undergoes extensive methylation changes over time and its contribution to T-cell development likely wanes.
A recent case series has outlined a multi-centre experience caring for 25 children and 22 adults with heterozygous FOXN1 mutations (Bosticardo et al. 2019). Of the children, 21 were identified following an abnormal NBS result, while 4 others were investigated for persistent severe lymphopenia and (or) recurrent infections. Most were clinically well except for viral respiratory infections, and were managed conservatively. Common non-infectious manifestations included eczema and nail dystrophy (not seen in our patient). Five patients displayed evidence of more profound lymphopenia and (or) severe infections, and 3 received a hematopoietic stem cell transplant for a clinical diagnosis of SCID, before their FOXN1 mutations had been identified. Unfortunately, transplant outcome has included persistent lymphopenia despite engraftment in 2 children, and mortality of infection in the third patient. With respect to adults included in the report, the majority did not report on any substantial recurrent infectious history; however, it is possible that individuals impacted by severe infections would not have survived into adulthood, thus de-facto excluding persons with more profound disease manifestations from the adult cohort. In terms of laboratory evaluation, the most common and consistent manifestation in infants and children was T-lymphopenia impacting both CD4+ and CD8+ populations, with improvement in CD4+ noted toward the second year of life. Among adult participants, CD8+ lymphopenia was the only persistent disease abnormality identified in some.
The spectrum of clinical manifestations and severity hitherto reported among children with heterozygous FOXN1 mutations has been broad, with no clear genotype-phenotype correlation with respect to location and nature of the mutations. Indeed, mutations reported by Bosticardo et al. (2019) spanned all gene domains, and included substitutions, insertions and deletions, in both exonic and intronic regions. However, the majority of cases involved missense mutations, while a splice site mutation was only reported in 1 child. It therefore remains to be determined whether splice-site variants result in a unique phenotype compared with other FOXN1 mutations. Importantly, while splice site mutations may be identified on whole exome sequencing given their proximity to exonic regions, deep intronic mutations would not be identified using this method, and would require either targeted sequencing of the FOXN1 gene in its entirety, or whole genome sequencing. Of note, deep intronic mutations have never been identified as causing heterozygous FOXN1 deficiency, but as whole genome sequencing becomes more prominently embedded in clinical practice, this observation may very well change.
In regards to management of children with heterozygous FOXN1 mutations, our centre has practiced conservative, yet diligent follow-up of any patient with abnormal lymphocyte counts and (or) function. In regards to live vaccines, our experience suggests that in children with FOXN1 mutations and normal T-cell function, whose T-lymphopenia is mild and improving or fully resolved, live viral vaccinations may be administered safely. However, a case-by-case discussion should be held with families, identified the potential risks and benefits of such an approach. Finally, genetic testing and counselling may be considered for asymptomatic first-degree family members as well.
REFERENCES
Bosticardo M., Yamazaki Y., Cowan J., Giardino G., Corsino C., Scalia G., Prencipe R., Ruffner M., Hill D.A., Sakovich I., and Yemialyanava I. 2019. Heterozygous FOXN1 variants cause low TRECs and severe T cell lymphopenia, revealing a crucial role of FOXN1 in supporting early thymopoiesis. Am. J. Hum. Genet. 105(3): 549–561.
Flanagan S.P. 1966. ‘Nude’, a new hairless gene with pleiotropic effects in the mouse. Genet. Res. 8(3): 295–309.
Frank J., Pignata C., Panteleyev A.A., Prowse D.M., Baden H., Weiner L., Gaetaniello L., Ahmad W., Pozzi N., Cserhalmi-Friedman P.B., and Aita V.M. 1999. Exposing the human nude phenotype. Nature, 398(6727): 473–474.
Lam E.W., Brosens J.J., Gomes A.R., and Koo C.Y. 2013. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer, 13(7): 482–495.
Nowell C.S., Bredenkamp N., Tetelin S., Jin X., Tischner C., Vaidya H., Sheridan J.M., Stenhouse F.H., Heussen R., Smith A.J., and Blackburn C.C. 2011. Foxn1 regulates lineage progression in cortical and medullary thymic epithelial cells but is dispensable for medullary sublineage divergence. PLoS Genet. 7(11): e1002348.
Pignata C., Fiore M., Guzzetta V., Castaldo A., Sebastio G., Porta F., and Guarino A. 1996. Congenital alopecia and nail dystrophy associated with severe functional T‐cell immunodeficiency in two sibs. Am. J. Med. Genet. 65(2): 167–170.
Romano R., Palamaro L., Fusco A., Giardino G., Gallo V., Del Vecchio L., and Pignata C. 2013. FOXN1: A master regulator gene of thymic epithelial development program. Front. Immunol. 4: 187.
Žuklys S., Handel A., Zhanybekova S., Govani F., Keller M., Maio S., Mayer C.E., Teh H.Y., Hafen K., Gallone G., and Barthlott T. 2016. Foxn1 regulates key target genes essential for T cell development in postnatal thymic epithelial cells. Nat. Immunol. 17(10): 1206–1215.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 1 • March 2021
Pages: 1 - 4
History
Received: 4 February 2021
Accepted: 25 February 2021
Accepted manuscript online: 26 February 2021
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
OriScott, JennyGarkaby, JessicaWillett-Pachul, Amarilla B.Mandola, DanieleMerico, and YehonatanPasternak. 2021. A novel splice site variant in FOXN1 in a patient with abnormal newborn screening for severe combined immunodeficiency and congenital lymphopenia. LymphoSign Journal.
8(1): 1-4. https://doi.org/10.14785/lymphosign-2021-0013
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Management of newborn screening for severe combined immunodeficiency at a quaternary referral centre—an updated algorithm
2. An unusual presentation of DiGeorge syndrome
3. Novel heterozygous FOXN1 mutation identified following newborn screening for severe combined immunodeficiency is associated with improving immune parameters
4. A novel mutation in TRAC in a patient with abnormal newborn screening for severe combined immunodeficiency
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
