Identification of a novel RAG1 hypomorphic mutation in a child presenting with disseminated vaccine-strain varicella
Abstract
Background: Recombination-activating gene 1 (RAG1) and recombination-activating gene 2 (RAG2) encode unique lymphocyte endonuclease proteins that are crucial in T and B cell development through V(D)J recombination. RAG1 gene defects lead to variable phenotypes, ranging from immunocompetent to severe combined immunodeficiency (SCID). Curative therapy for severe manifestations can be achieved through hematopoietic stem cell transplantation (HSCT). Advances in genomic sequencing have led to the discovery of new variants and it is recognized that the level of recombinase activity correlates with disease severity.
Aim: To report the clinical presentation, immunological work-up, decision process to undergo HSCT, and confirmatory genetic diagnosis in a patient who was well until her initial presentation with disseminated vaccine-strain varicella.
Methods: Clinical data was gathered through retrospective chart review. Immunological investigations, targeted gene sequencing, and thymic biopsy results were reviewed. Further genetic analysis, including whole exome and whole genome sequencing was performed.
Results: Whole exome sequencing identified a single missense mutation in RAG1, R474C (c.1420C>T), which would not account for the clinical presentation. Healthy individuals with only 1 mutation have been reported. Subsequently, whole genome sequencing revealed a novel second heterozygous missense variant, H945D (c.2833G>T) in the RAG1 gene.
Conclusion: Hypomorphic RAG1 mutations with residual activity have a diverse phenotypic expression. Identifying and understanding the implications of these mutations is crucial for disease prognostication and tailoring management.
Statement of novelty: We present a novel RAG1 missense variant, with likely complete or partial loss of function, in a patient with significant impairment in cellular immunity.
Introduction
Recombination-activating gene 1 (RAG1) and recombination-activating gene 2 (RAG2) encode unique lymphocyte endonuclease proteins that form the complex required for somatic V(D)J gene recombination (Fugmann 2001). This process generates receptor diversity—equipping T and B cells with a broad repertoire for antigen recognition. DNA is cleaved at recombination signal sequences, resulting in hairpin-capped double-strand breaks that are opened and processed by Artemis (Sadofsky 2001). The ends are eventually joined by non-homologous end joining with DNA ligase (Notarangelo et al. 2016). RAG1 may also facilitate the subsequent joining of the coding ends (Notarangelo et al. 2016; Delmonte et al. 2018). T and B cell development is dependent on this process and cannot progress beyond the DN3 and pre-B-1 stage without RAG1 or RAG2 (Gennery 2019). The discarded DNA from thymic T-cell rearrangement forms a loop known as a T-cell receptor excision circle (TREC), which is now used in newborn screening (NBS) for severe combined immunodeficiency (SCID) (Chitty-Lopez et al. 2020).
There is a recognized spectrum of phenotypes which correlates with the level of recombinase activity (Delmonte et al. 2018; Gennery 2019). Null mutations give rise to a T-B-NK+ SCID phenotype, whereas hypomorphic missense mutations with residual RAG activity can be found in Omenn syndrome (Gennery 2019). RAG mutations have also been implicated in cases of combined immunodeficiencies, common variable immunodeficiency (CVID), antibody deficiencies, granulomatous disease, and autoimmunity (Delmonte et al. 2018). We report a unique case of a patient with compound heterozygous RAG1 variants, who was well until her initial presentation with vaccine-strain varicella.
Methods
Chart review
Patient data was gathered by retrospective chart review. This included immunological investigations pre- and post-transplant, targeted gene sequencing results, thymic biopsy findings, and clinical documentation. Informed consent was obtained from the family in accordance with the SickKids Research Ethics Board (Protocol No. 1000005598).
Genetic analysis
DNA from blood was submitted for analysis at the TCAG, Hospital for Sick Children, Toronto, Ontario. For whole exome sequencing (WES), 100 ng of input DNA was utilized for library preparation (Ion AmpliSeq Exome Kit; Life Technologies). The exome library was subsequently immobilized on Ion PI™ Ion Sphere particles (Ion PI Template OT2 200 Kit v3; Life Technologies) and sequenced on the Ion PI™ Sequencing 200 Kit v3 and Ion PITM Chip v2 in the Ion Proton semiconductor sequencing system (Life Technologies). Alignment and variant calling were performed with Torrent Suite (v4.0) on the Ion Proton Server using the Ion Proton AmpliSeq germline low-stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline 35 (based on Annovar), 36 and RefSeq gene models (downloaded from UCSC). For whole genome sequencing (WGS), the library was prepared using 700 ng of genomic DNA and sequenced on 1 lane (average depth of 37X) of the Illumina HiSeq X platform. Reads were aligned using BWA mem v0.7.12 to GRCh37; single nucleotide variants (SNVs) and insertions/deletions (INDELs) were called using GATK 3.7 haplotype caller. Copy number variants (CNV), comprising losses and gains with size ≥1 kb, were called using a pipeline based on the read depth callers ERDS and CNVnator. Variants were prioritized based on variant quality, allele frequency, molecular effect, gene function, and phenotype. Genomic coordinates are based on hg19/build37. Gene product effects were reported, unless otherwise indicated, in relation to the RefSeq transcript predicted as principal by APPRIS4. Only SNVs and INDELs with GATK VQSR filter PASS were considered for further analysis. Allele frequencies of SNVs and INDELs were obtained from gnomAD and ExAC. Missense impact predictions were obtained from dbNSFP for SIFT, PolyPhen2, Mutation Assessor and CADD. Splicing impact predictions were obtained from SPIDEX and dbscSNV.
Results
Clinical features
The patient was born at term following an uneventful pregnancy. She was infection-free with no concerns for growth, development, autoimmunity, or atopy until she received her live vaccines. These consisted of measles, mumps, rubella, and varicella at the age of 1. Within 2 weeks she developed fever and a vesicular rash, suspected to be varicella, which progressed to purpura of the limbs. Given the similarities between her and a sibling who had passed away, she was admitted to the intensive care unit and treated aggressively with immunosuppressants, steroids, and antivirals. Renal biopsy was normal, with no markers of collagen vascular disease. Skin biopsy was consistent with small vessel vasculopathy and microthrombosis. Her bone marrow had decreased lymphocytes but no malignant cells. Vaccine-strain varicella was isolated from skin lesions and cerebral spinal fluid, although there was no evidence of central nervous system or internal organ involvement. Treatment was advanced to include infliximab and rituximab, in conjunction with steroids and cyclosporin, with good clinical response. She was discharged home to continue intravenous immunoglobulin (IVIG), immunosuppressants, prophylactic antibiotics, and antivirals. Physical examination did not reveal any dysmorphisms, organomegaly, or erythroderma. Small lymph nodes were palpable; she had a normal cardiorespiratory and musculoskeletal exam.
Following discharge, she developed diarrhea and failure to thrive (FTT), necessitating nasogastric tube feeding. She was given a provisional diagnosis of celiac disease. The patient was also found to be positive for norovirus and C. difficile, which was treated. There was mild eczema that was well controlled with topical hydrocortisone and moisturizers. She experienced 1 episode of pneumonia around the age of 2, which responded to oral antibiotics. Her immune work-up was initially complicated due to her intercurrent illness, immunosuppressants, and IVIG. Following discontinuation of her medications, she continued to demonstrate impaired T-cell functioning with poor phytohemagglutinin (PHA) response, undetectable TRECs, abnormal expansion of TCR-Vbeta repertoire, and an abnormal thymic biopsy. There were also impaired antibody responses.
Family history
Parents were non-consanguineous and healthy. Three years prior to our patient’s presentation, they had given birth to fraternal twins. The boy was healthy. The girl developed a severe vasculitic rash prior to the age of 1 and was hospitalized. Biopsy of the rash was concerning for infantile polyarthritis nodosa. She had decreased C3 and C4 with hypergammaglobulinemia. She developed several line-related infections during her hospitalization. Despite treatment with IVIG, steroids, and cyclophosphamide, she passed away at the age of 2 from gastrointestinal and pulmonary vasculitis, as well as renal failure. Autopsy revealed minimal germinal follicles in her lymph nodes. The germinal centres and paracortex lacked well-formed secondary follicles and were mainly composed of primary lymphoid follicles. However, it was difficult to discern if this was a primary finding or secondary to her illness and immunosuppression.
Immunological investigations pre-transplant
The patient was initially seen by Immunology as an inpatient. However, at that time immune work-up could have been complicated by her acute illness and multiple therapies. After cessation of IVIG and immunosuppressants, initial immunological work-up revealed reduced WBC of 2.8 × 109/L and lymphocytes of 1.12 × 109/L. Immunoglobulins were normal (IgG 8.9, IgA 0.9, IgM 0.6). Isohemagglutinin anti-B was negative. Lymphocyte immunophenotyping revealed CD4+364 (normal 1573–2949), CD8+ 76 (normal 472–1107), CD16+56+ 412 (normal 155–565), and CD19+ 178 (normal 434–1274). Lymphocyte proliferation test was significantly decreased at 8% of control (stimulation index 40/455). TRECs were undetectable. TCR-Vbeta repertoire was abnormal, with expansion in Vbeta7, Vbeta24, Vbeta3, and Vbeta14 clones, accompanied by underrepresentation of other TCR clones. There was lack of a CD45RA naïve population. Targeted gene sequencing did not reveal a genetic diagnosis. A thymic biopsy showed absence of Hassall’s corpuscles and no clear corticomedullary distinction.
Management
The patient had evidence of severe cellular impairment on immunological work-up. Given these findings, in conjunction with her FTT, thymic dysplasia, infection history, and sibling’s course, the decision was made to proceed with bone marrow transplant (BMT). This was reached after careful deliberation in consultation with other experts and the family, as she did not yet have a genetic diagnosis at that time. She underwent a matched sibling donor BMT at the age of 2. Mild skin graft-versus-host disease occurred during her cyclosporin wean but she otherwise tolerated the procedure well. She was slowly weaned off her immunosuppressants; her IVIG and antibiotic prophylaxis were subsequently stopped. She had full engraftment with good evidence of immune reconstitution. Two years post-transplant the patient received her killed vaccines. The following year, she tolerated her live-viral vaccines. She developed 1 episode of Streptococcus pneumoniae bacteremia and a few episodes of pneumonia between the ages of 6–9, which resolved after initiation of chest physiotherapy and a steroid inhaler.
Immunological investigations post-transplant
Post-transplant, full engraftment with 100% donor chimerism was achieved. Her most recent evaluation reveals a normal complete blood count with WBC of 4.37 × 109/L and lymphocytes of 2.0 × 109/L. Normal immunoglobulins (IgG 10, IgA 1.3 and IgM 0.9). Isohemagglutinin anti-B was 1:8. Vaccine titres were protective, including varicella. Lymphocyte immunophenotyping was all within normal range (CD4+ 916, CD8+ 443, CD16+56+ 279, CD19 314). Lymphocyte proliferation was robust with a stimulation index of 605. TCR-Vbeta revealed slight expansion of Vbeta5 and borderline low representation of Vbeta9—there was good representation of all other families. Furthermore, she had appropriate numbers of both naïve and memory T-cells.
Genetic work-up
Around the time of her initial presentation, targeted sequencing of CD3delta, AIRE, and ReIB were negative. Whole exome sequencing in 2014 identified a single mutation in RAG1; however, this was deemed insufficient to explain the phenotypic presentation.
Subsequently, in 2020 WGS revealed 2 separate heterozygous missense variants in the RAG1 gene (NM_000448): R474C (c.1420C>T) and H945D (c.2833 G>T) (Table 1). Sanger sequencing of parental DNA revealed the mother to be heterozygous for the R474C variant and the father was heterozygous for the H945D variant.
Table 1:

Discussion and conclusions
We present a case of a child with a concerning family history who presented after her first live-viral vaccinations and was found to have severe T-cell dysfunction. Live-viral vaccines are contraindicated in individuals with severe impairment of cellular immunity. Several cases of vaccine-strain illnesses, including varicella, have been reported in immunodeficient patients (Bayer et al. 2014; Leung et al. 2014). With the available testing at the time, our patient presented a diagnostic and therapeutic dilemma as there was no genetic diagnosis. However, her family history pressed upon the gravity of a timely decision, which was made following deliberation and an abnormal thymic biopsy. Successful immune reconstitution was achieved through BMT. Diagnostic confirmation of 2 RAG1 variants was found several years later, with advancements in genomic testing through WGS.
RAG1 gene defects lead to variable phenotypes, some of which are yet to be discovered or described. SCID is characterized by life-threatening infections in early infancy due to absent or minimal T-cell numbers and functioning; mutations in RAG1 account for approximately 4% of cases (Delmonte et al. 2018). Omenn syndrome is the result of hypomorphic mutations that allow for occasional recombination events (Shearer et al. 2014). Individuals present in early infancy with erythroderma, lymphadenopathy, and eosinophilia due to autoreactive oligoclonal T-cell expansion that infiltrates the skin, gut, liver, and other organs (Delmonte et al. 2018). Atypical SCID presents with a severe rash but lacks the organomegaly from lymphoproliferation (Delmonte et al. 2018). Our patient presented later in life and did not display erythroderma, lymphadenopathy, or organomegaly. She did have manifestations of autoimmunity and responded to immunosuppression and immunomodulation. In 2008, a report of 3 unrelated females who presented later in life with granulomas, severe viral infections, and B-cell lymphoma highlighted RAG variants who present later with autoimmunity and inflammation (Schuetz et al. 2008). The frequency of RAG1/2 hypomorphic mutations in the general population is higher than previously thought. Kumánovics et al. (2017) estimated a population frequency of up to 1:181 000 and suggested mutations likely contribute to undiagnosed cases of combined immunodeficiencies.
The RAG1 gene is located on chromosome 11p12; it has 1 protein-coding exon and is composed of 1043 amino acids (Notarangelo et al. 2016). Two missense RAG1 variants were identified in our patient, R474C and H945D, and are likely causal given her phenotypic presentation (Figure 1). Furthermore, both mutations occur in the catalytic core of human RAG1, which is comprised of amino acids 387–1011 (Notarangelo et al. 2016).
Figure 1:
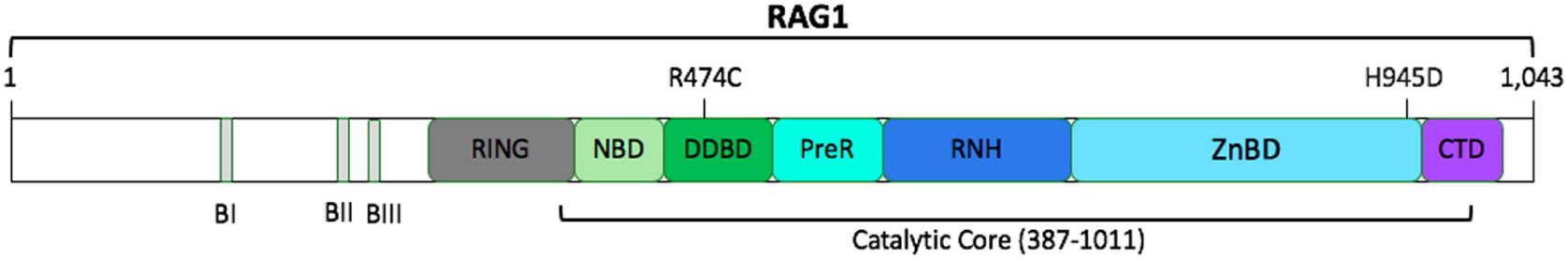
The R474C mutation has previously been reported in cases of SCID, Omenn syndrome, and CD4+ T-cell lymphopenia (Chen et al. 2014). Schönberger et al. (2009) published a case of a 3-month-old girl with Omenn syndrome who successfully underwent a matched cord transplant. She had a compound heterozygous defect within the RAG1 gene, R474C and R975W. A 2-year-old girl with recurrent, severe cavitating pneumonias and herpes zoster infections was found to have CD4 T-cell lymphopenia (Avila et al. 2010). Mutational analysis of RAG1 showed compound heterozygosity with R474C (c.1420C>T) and K86fs (c.256-7 DelAA) (Avila et al. 2010). Engraftment was achieved following a matched unrelated BMT. Kuijpers et al. (2011) identified 2 missense mutations in RAG1, R474C) and L506F. The child presented with hemorrhagic cutaneous skin lesions and varicella-associated pneumonia, bronchiectasis, reduced naïve lymphocytes and low TRECs (Kuijpers et al. 2011). Interestingly, proliferation capacity was intact and antibody titres to vaccines were protective (Kuijpers et al. 2011). The R474C recombinant protein was found to have 25% recombinase activity compared to wild type, while the L506F had no activity (Kuijpers et al. 2011). Chen et al. (2014) described a family with 2 phenotypically affected children, each with the compound heterozygous mutations c.1420C>T and c.2949delA. Both developed vaccine-strain varicella from a vaccinated family contact, autoimmune cytopenias, and reduced naïve CD4+ T-cell numbers (Chen et al. 2014). Sanger sequencing revealed that the parents were each a carrier of 1 mutation and there were 2 unaffected siblings with the R474C (c.1420C>T) mutation (Chen et al. 2014).
The H945D (c.2833G>T) variant in our patient is a novel mutation that has not been previously described in the literature. Individuals carrying a single R474C (c.1420C>T) mutation have been found to be healthy; only when a second mutation with similar or further reduced recombinase activity is present, does it lead to clinical sequalae (Chen et al. 2014). Thus, the H945D mutation results in either complete or partial loss of function and it is the combination of both mutations that is responsible for our patient’s presentation. This is further evident as her clinically healthy parents carry one of each mutation—with the mother being a carrier for the R474C variant and the father for the H945D variant. Consequently, it is pertinent to consider compound heterozygous RAG1 mutations in individuals with impaired cellular immunity, autoimmunity, and lymphopenia.
With the introduction of TREC quantification on the NBS, this may lead to earlier detection of individuals with RAG1 hypomorphic mutations. For our patient, SCID NBS had not yet been implemented. However, TREC levels measured after her acute presentation were undetectable. Although we are unable to definitively conclude if she would have been picked up on NBS, it can be postulated that she could have had abnormal TREC levels from birth given her impaired T-cell function. As the second H945D mutation was only identified after improved detection capabilities onWGS, it would be important to consider employing such methods of work-up when conventional genetic panels do not yield findings. Early HSCT following detection of pathogenic variants is postulated to circumvent autoimmune complications and the development of significant infections (Chen et al. 2014). However, given the diversity of disease severity, ongoing efforts to correlate genotype with phenotype will help with understanding the implication of novel variants. In the absence of genetic confirmation, consideration of clinical progression, immunologic work-up, and family history is warranted. Diagnostic tests and management strategies should be selected accordingly.
REFERENCES
Avila E.M., Uzel G., Hsu A., Milner J.D., Turner M.L., Pittaluga S., Freeman A.F., and Holland S.M. 2010. Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics, 126(5): e1248–e1252.
Bayer D.K., Martinez C.A., Sorte H.S., Forbes L.R., Demmler‐Harrison G.J., Hanson I.C., Pearson N.M., Noroski L.M., Zaki S.R., Bellini W.J., and Leduc M.S. 2014. Vaccine‐associated varicella and rubella infections in severe combined immunodeficiency with isolated CD4 lymphocytopenia and mutations in IL7R detected by tandem whole exome sequencing and chromosomal microarray. Clin. Exp. Immunol. 178(3): 459–469.
Chen K., Wu W., Mathew D., Zhang Y., Browne S.K., Rosen L.B., McManus M.P., Pulsipher M.A., Yandell M., Bohnsack J.F., and Jorde L.B. 2014. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J. Allergy Clin. Immunol. 133(3): 880–882.e10.
Chitty-Lopez M., Westermann-Clark E., Dawson I., Ujhazi B., Csomos K., Dobbs K., Le K., Yamazaki Y., Sadighi Akha A.A., Chellapandian D., and Oshrine B. 2020. Asymptomatic infant with atypical SCID and novel hypomorphic RAG variant identified by newborn screening: A diagnostic and treatment dilemma. Front. Immunol. 11: 1954.
Delmonte O.M., Schuetz C., and Notarangelo L.D. 2018. RAG deficiency: Two genes, many diseases. J. Clin. Immunol. 38(6): 646–655.
Fugmann S.D. 2001. RAG1 and RAG2 in V(D)J recombination and transposition. Immunol. Res. 23(1): 23–39.
Gennery A. 2019. Recent advances in understanding RAG deficiencies. F1000Res. 8: 148.
Kuijpers T.W., IJspeert H., van Leeuwen E.M., Jansen M.H., Hazenberg M.D., Weijer K.C., Van Lier R.A., and van der Burg M. 2011. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood, 117(22): 5892–5896.
Kumánovics A., Lee Y.N., Close D.W., Coonrod E.M., Ujhazi B., Chen K., MacArthur D.G., Krivan G., Notarangelo L.D., and Walter J.E. 2017. Estimated disease incidence of RAG1/2 mutations: A case report and querying the Exome Aggregation Consortium. J. Allergy Clin. Immunol. 139(2): 690–692.e3.
Leung J., Siegel S., Jones J.F., Schulte C., Blog D., Scott Schmid D., Bialek S.R., and Marin M. 2014. Fatal varicella due to the vaccine-strain varicella-zoster virus. Hum. Vaccin. Immunother. 10(1): 146–149.
Notarangelo L.D., Kim M.S., Walter J.E., and Lee Y.N. 2016. Human RAG mutations: Biochemistry and clinical implications. Nat. Rev. Immunol. 16(4): 234–246.
Sadofsky M.J. 2001. The RAG proteins in V(D)J recombination: More than just a nuclease. Nucleic Acids Res. 29(7): 1399–1409.
Schönberger S., Ott H., Gudowius S., Wüller S., Baron J.M., Merk H.F., Lassay L., Megahed M., Feyen O., Laws H.J., and Dilloo D. 2009. Saving the red baby: Successful allogeneic cord blood transplantation in Omenn syndrome. Clin. Immunol. 130(3): 259–263.
Schuetz C., Huck K., Gudowius S., Megahed M., Feyen O., Hubner B., Schneider D.T., Manfras B., Pannicke U., Willemze R., and Knüchel R. 2008. An immunodeficiency disease with RAG mutations and granulomas. N. Engl. J. Med. 358(19): 2030–2038.
Shearer W.T., Dunn E., Notarangelo L.D., Dvorak C.C., Puck J.M., Logan B.R., Griffith L.M., Kohn D.B., O’Reilly R.J., Fleisher T.A., and Pai S.Y. 2014. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: The primary immune deficiency treatment consortium experience. J. Allergy Clin. Immunol. 133(4): 1092–1098.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 8 • Number 1 • March 2021
Pages: 5 - 10
History
Received: 6 February 2021
Accepted: 19 February 2021
Accepted manuscript online: 21 February 2021
Notes
There are no conflicts or funding sources to declare.
Copyright
© 2021.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MeiXu, BrendaReid, DanieleMerico, and Chaim M.Roifman. 2021. Identification of a novel RAG1 hypomorphic mutation in a child presenting with disseminated vaccine-strain varicella. LymphoSign Journal.
8(1): 5-10. https://doi.org/10.14785/lymphosign-2021-0014
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
