Histopathological features of patients with chronic granulomatous disease
Abstract
Introduction: Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by defects in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. Affected patients suffer recurrent life-threatening infections due to phagocyte dysfunction and dysregulation of the immune system. Histopathological assessment is important to help identify the extent and severity of infection and tissue injury.
Aim: We present pathological findings in 5 patients with CGD who were followed at our centre.
Methods: Patient information was reviewed retrospectively in accordance with local institutional guidelines. All patients had confirmed diagnosis of CGD with mutation in one of the 5 subunits of the NADPH oxidase.
Results: Histopathological features of the gastrointestinal tract, liver, and spleen are noted, and include the presence of granulomatous inflammation and pigmented macrophages.
Discussion: It is essential for clinicians to keep primary immunodeficiency as one of the differential diagnoses in patients with severe infection or inflammation, whether in the absence or presence of granuloma formation. The detection of PAS-positive macrophages, diffuse granulomatous inflammation, and hepatic abscesses should raise strong suspicion of CGD.
Statement of novelty: We describe the histopathological findings of a paediatric cohort of patients with CGD.
Background
Chronic granulomatous disease (CGD) is a primary immunodeficiency that affects an estimated 1:200 000 live births (Holland 2010). It is caused by defects in the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which is required for respiratory burst activity and killing of microorganisms (Roos et al. 2003). Thus, while phagocytes are able to ingest invading microorganisms, in the absence of respiratory burst, they are unable to kill them. Mutations in any one of the 5 genes which encode the complex can result in partial or complete ablation of function (Kuhns et al. 2010). These include the membrane localized gp91phox and p22phox, encoded by CYBB and CYBA, respectively, as well as the cytosolic components p47phox, p67phox, and p40phox encoded by NCF1, NCF2, and NCF4, respectively. Defects in gp91phox result in X-linked inheritance and affects an estimated 65% of cases in North America (Winkelstein et al. 2000), whereas mutations in p22phox, p47phox and p40phox are passed on in an autosomal recessive manner. Rarely, X-linked inheritance of CGD is found in females due to lyonization (Roos and de Boer 2014; Marciano et al. 2018).
Patients with CGD usually present within the first 3 years of life with difficult to treat recurrent bacterial and fungal infections (Thomsen et al. 2016), especially from catalase-positive organisms which degrade hydrogen peroxide and further hinder the microbicidal activity of affected phagocytes (neutrophils, monocytes, macrophages, eosinophils) (Ben-Ari et al. 2012). Infections are commonly associated with the bacteria Staphylococcus aureus, Serratia mercescens, Burkholderia cepacia, Nocardia, Streptococcus, Salmonella species, and Klebsiella species, while Aspergillus fumigatus and Candida albicans are the leading fungal culprits (Ben-Ari et al. 2012; Falcone et al. 2012). As a result of the recurrent infections, patients with CGD develop lymphadenitis, abscesses, and granuloma formation. Other manifestations include osteomyelitis, purulent dermatitis, and cellulitis.
Histopathology is an important aspect of the work-up for CGD, assisting to determine the composition of cellular infiltrates, extent and severity of tissue injury, as well as identity of invading bacterial and fungal organisms. Various organs may be affected, including the gastrointestinal tract, liver, skin and soft tissue, lung, bone, bladder, and lymphoreticular systems.
Non-caseating (non-necrotizing) granuloma formation is a hallmark of CGD and may appear in the lungs, liver, spleen, gastrointestinal tract, urinary tract, and skin (Schappi et al. 2008). Non-specific abscess formation or acute inflammation is often present, alongside accumulated pigmented macrophages (Levine et al. 2005). While histopathology alone is insufficient to provide unequivocal indication of CGD, it can be used to raise the suspicion of disease.
Here, we present histopathological findings from 5 paediatric patients with confirmed diagnosis of CGD.
Methods and results
Patient informed consent was obtained and charts were retrospectively reviewed in accordance with institutional protocols. All patients had a confirmed clinical diagnosis of CGD between 2008 and 2018 at the Hospital for Sick Children, Toronto, ON.
Patient 1
Splenic biopsy from a 7 year old female detected diffuse infiltrate with small, well-defined non-caseating granulomas (Figure 1). The patient presented with fatigue, reduced appetite, and abdominal pain associated with splenomegaly and hepatomegaly. A diagnosis of CGD was confirmed following the identification of a mutation in NCF1.
Figure 1:
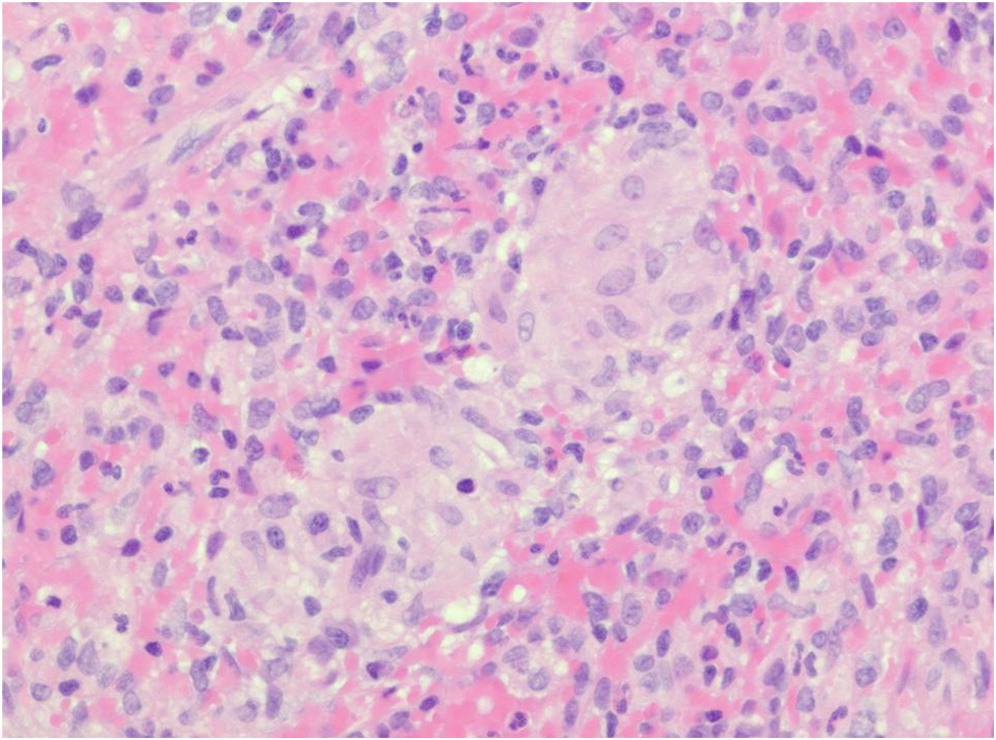
Patient 2
Liver biopsy from an 18 month old male showed necrotizing granulomatous inflammation and the presence of epithelioid histiocytes (Figure 2). Hypoechoic lesions in the liver were also identified during radiography and Serratia marcescens detected by urine culture. The patient suffered severe colitis and together with repeat low neutrophil oxidative burst index test results, the suspicion of CGD was raised. Genetic testing later revealed a mutation in CYBB, causing X-linked CGD. The patient underwent sibling donor hematopoietic stem cell transplantation, however developed various issues post-transplant, including reactivation of colitis and plasmacytoma of his tongue (Figure 3).
Figure 2:
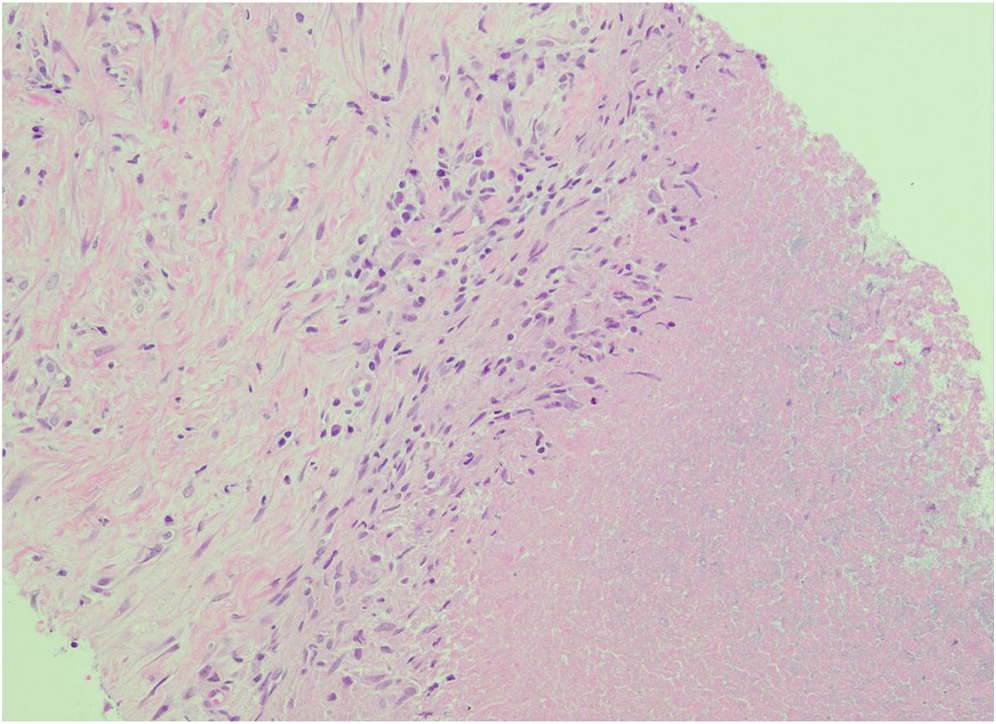
Figure 3:
Patient 3
GI biopsies from a 3 year old male with X-linked CGD due to mutation in CYBB revealed focal eosinophilic abscess in the duodenum but no granulomas. Multiple pigmented macrophages were found in the lamina propria of sigmoid colon and rectum (Figure 4A). These macrophages were positive for Periodic acid-Schiff (PAS) staining (Figure 4B). Radiographic and endoscopic investigation of the GI tract as well as upper and lower GI endoscopy were initiated following significant episodes of vomiting, abdominal pain, and obstruction. Diffuse inflammation was present from the esophagus to small bowel.
Figure 4:
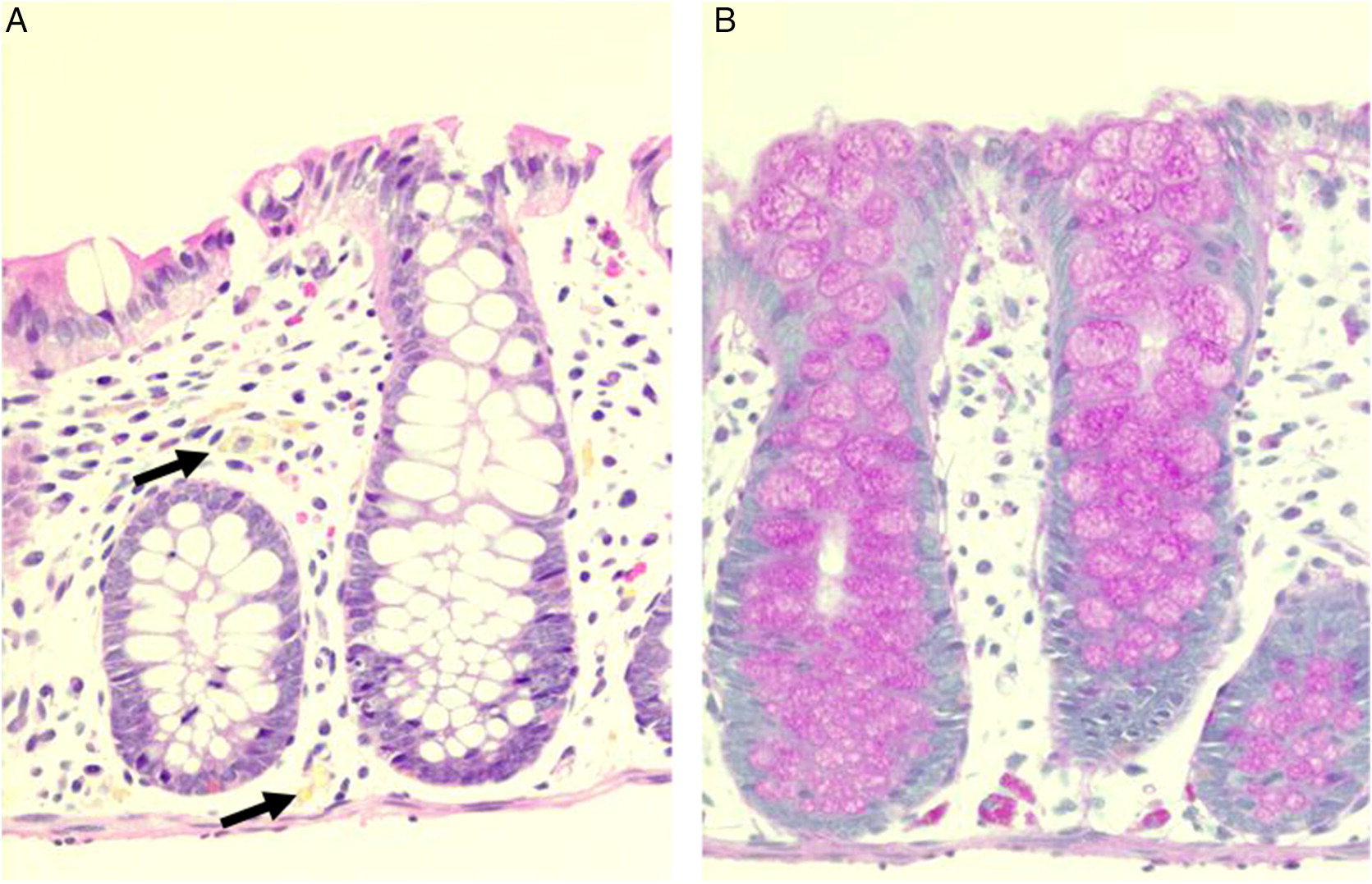
Patient 4
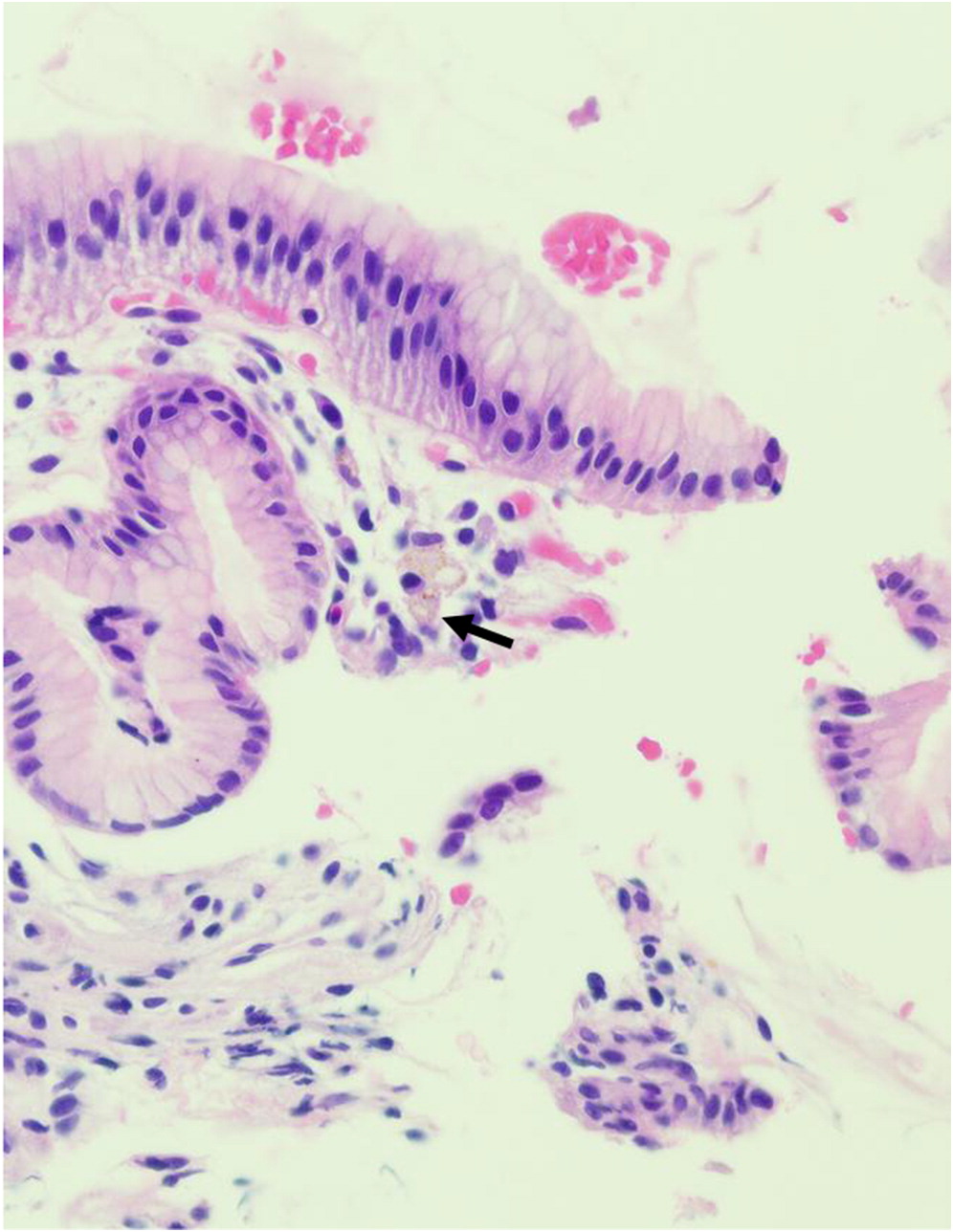
Gastric antrum biopsy from an 18 month old male, presenting with perianal abscess and fistula formation, revealed scattered pigmented macrophages throughout the stomach (Figure 5) and colon. The patient developed mouth ulcers that would wax and wane every month, raising the suspicion of CGD. Genetic testing confirmed mutation within CYBB, consistent with X-linked CGD.
Figure 5:
Patient 5
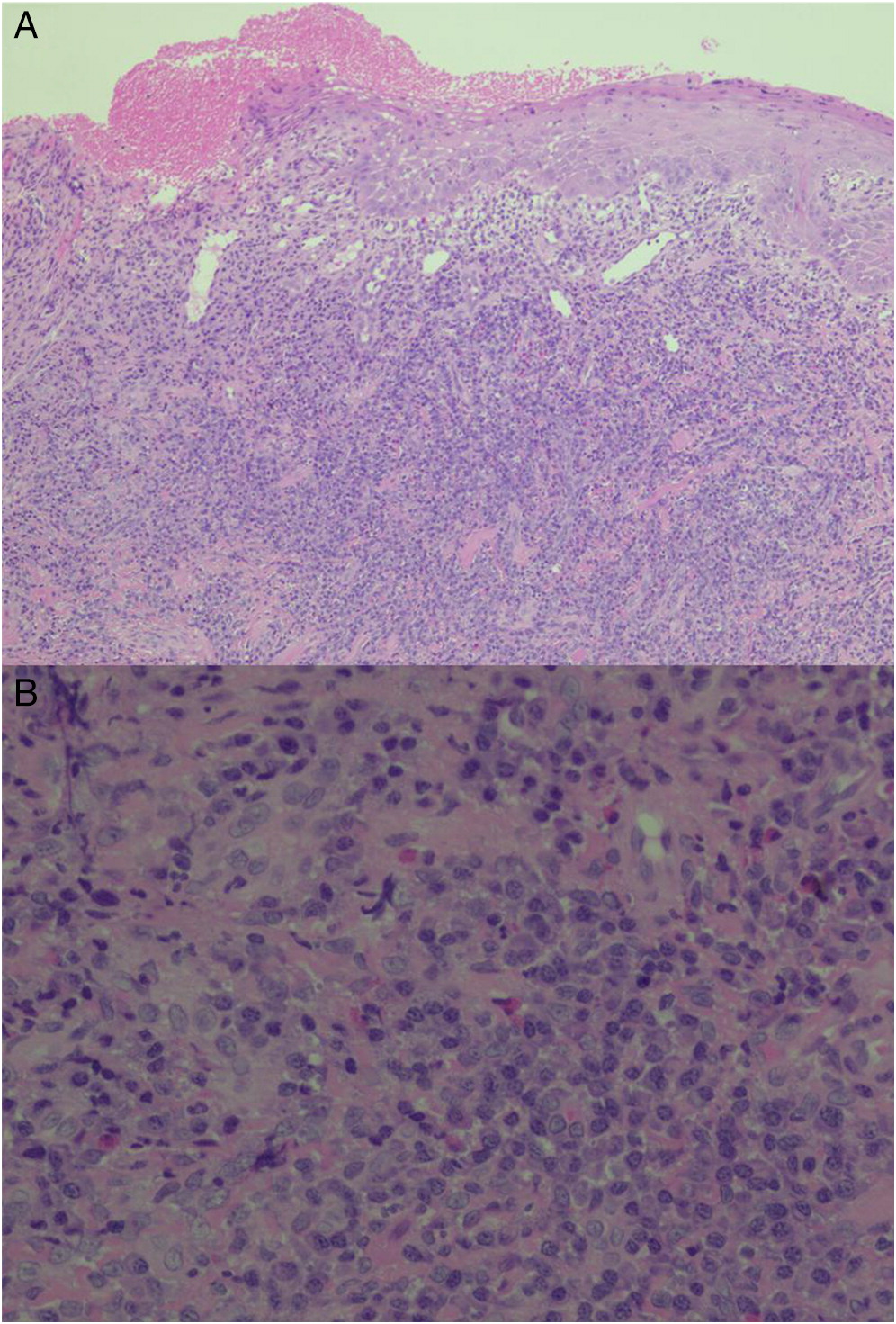
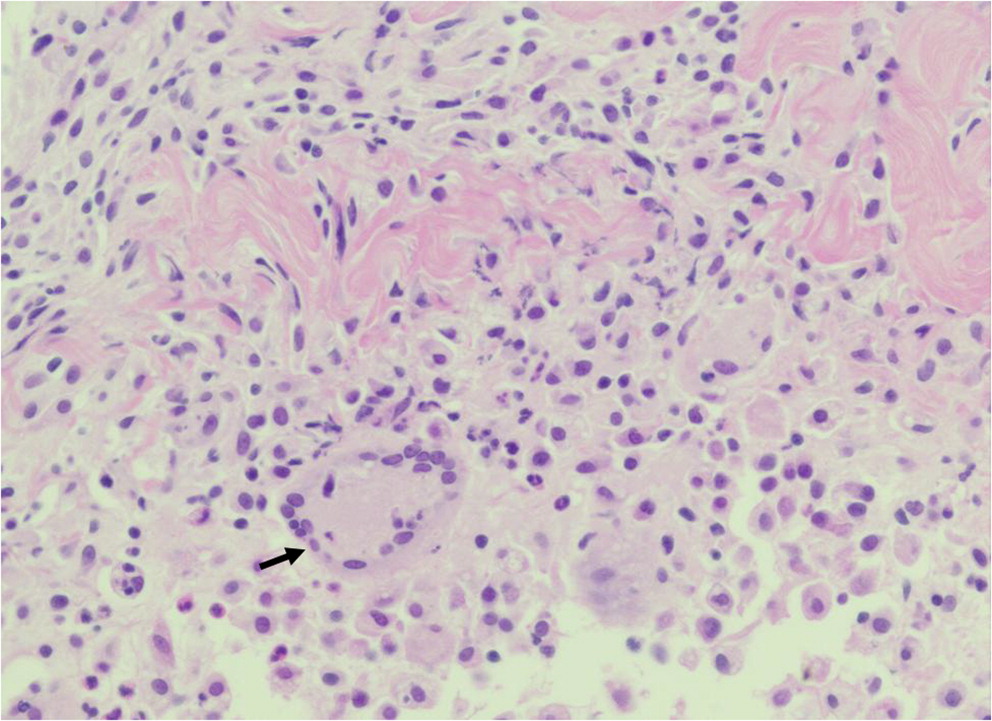
Skin biopsy from the thigh of a 15 year old male with X-linked CGD associated with CYBB mutation identified granulomatous inflammation, including clusters of epithelioid histiocytes and giant cells (Figure 6). The patient presented with lymphadenopathy and abscesses in his right foot, thigh, and lung. No infectious etiology was identified.
Figure 6:
Discussion
Infections in patients with CGD are often associated with organs that are exposed to the external environment—including skin, lungs, and GI tract, as well as the lymph nodes where foreign antigens are processed. Furthermore, dissemination of microorganisms can result in liver abscesses and osteomyelitis. The results of histopathological findings can raise the suspicion of CGD and help to distinguish it from other childhood diseases that may affect similar organ systems. While there are often common findings of non-specific acute and (or) chronic inflammation, granulomatous inflammation is identified in up to 40% of cases (Levine et al. 2005). Granuloma formation is a classic feature of CGD, and is thought to arise from the immune dysregulation associated with ineffective phagocyte killing and persistent recruitment of macrophages. Hyperinflammation and autoimmune phenomena is common in CGD patients, and has been attributed to abrogated anti-inflammatory signaling. Often, pigmented macrophages containing inadequately digested intracellular debris are also present in affected tissues. Here, we highlight the histopathological findings in 5 patients with genetically confirmed CGD.
An estimated 90% of patients with CGD have hepatosplenomegaly, and abscesses in the spleen or liver may also occur (Zimmermann 2016). Splenic biopsy from patient 1 identified the presence of non-caseating granulomas. In the absence of other infectious etiology, a diagnosis of juvenile sarcoidosis was made. However, subsequent repeat low neutrophil oxidative burst indices raised the suspicion of CGD, and genetic testing later confirmed an autosomal recessive mutation in NCF1. Necrotizing granulomatous inflammation was also identified in the liver biopsy of patient 2. Bacterial hepatic abscesses are a hallmark of CGD (Chusid 1978; Lublin et al. 2002; Szekely et al. 2012), and have been reported in approximately 30% of patients (largely associated with Staphylococcus aureus infection) (Winkelstein et al. 2000). Microabscesses in the liver and spleen can also develop as a result of fungemia. Due to the rarity of hepatic abscesses in young children (Lublin et al. 2002), its detection should trigger investigation of CGD as a potential cause.
Manifestations of CGD in the GI tract can mimic other inflammatory bowel diseases (Rieber et al. 2012), affecting up to 50% of CGD patients and may precede diagnosis of the disease (Winkelstein et al. 2000; van den Berg et al. 2009). Clinical symptoms include vomiting, diarrhea, abdominal pain, and failure to thrive (Marciano et al. 2004), and is often misdiagnosed as very early onset inflammatory bowel disease in young children (Harris and Boles 1973). Histopathological evaluation may reveal inflammatory lesions anywhere along the length of the GI tract, from the mouth to the anus, with or without infectious etiology (Huang et al. 2006; Towbin and Chaves 2010). Often, biopsies reveal granuloma formation, nuclear debris, as well as the presence of pigmented macrophages in the lamina propria. Localization of pigmented macrophages in the deep layers of tissue, from the lamina propria through to the mucosa, submucosa and muscularis propria, has also been reported in GI biopsies of affected patients (Alimchandani et al. 2013). In addition, eosinophilia and microscopic eosinophilic crypt abscesses are usually detected in the absence of acute, neutrophilic inflammation. In patient 3, eosinophilic crypt abscesses and PAS positive macrophages were identified in the duodenum and rectum, respectively. His vomiting was explained by gastric outlet obstruction—part of a spectrum of manifestations of CGD which include colitis, dysmotility, abscesses, strictures, and oral ulcers (Marciano et al. 2004; Damen et al. 2010). In patient 4, scattered pigmented macrophages were identified in biopsies of the stomach and colon, collected during upper and lower GI endoscopy.
Involvement of the skin has been reported in up to 65% of CGD patients (Dohil et al. 1997), and include inflammatory lesions associated with aspergillosis or without infectious etiology, cutaneous granulomas, and on some occasions the presence of pigmented macrophages (Levine et al. 2005). The inflammatory process/abscess identified in the foot and thigh of patient 5 is an uncommon finding and rarely reported.
In summary, we show that histopathology in combination with careful assessment of patient history, radiography, and phagocyte cell function analysis, can raise strong suspicion of CGD.
REFERENCES
Alimchandani M., Lai J.P., Aung P.P., Khangura S., Kamal N., Gallin J.I., Holland S.M., Malech H.L., Heller T., Miettinen M., and Quezado M.M. 2013. Gastrointestinal histopathology in chronic granulomatous disease: A study of 87 patients. Am. J. Surg. Pathol. 37:1365–1372.
Ben-Ari J., Wolach O., Gavrieli R., and Wolach B. 2012. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti-Infect. Ther. 10:881–894.
Chusid M.J. 1978. Pyogenic hepatic abscess in infancy and childhood. Pediatrics. 62:554–559.
Damen G.M., van Krieken J.H., Hoppenreijs E., van Os E., Tolboom J.J., Warris A., Yntema J.B., Nieuwenhuis E.E., and Escher J.C. 2010. Overlap, common features, and essential differences in pediatric granulomatous inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 51:690–697.
Dohil M., Prendiville J.S., Crawford R.I., and Speert D.P. 1997. Cutaneous manifestations of chronic granulomatous disease. A report of four cases and review of the literature. J. Am. Acad. Dermatol. 36:899–907.
Falcone E.L., Hanses S., Stock F., Holland S.M., Zelazny A.M., and Uzel G. 2012. Streptococcal infections in patients with chronic granulomatous disease: Case report and review of the literature. J. Clin. Immunol. 32:649–652.
Harris B.H. and Boles E.T. Jr. 1973. Intestinal lesions in chronic granulomatous disease of childhood. J. Pediatr. Surg. 8:955–956.
Holland S.M. 2010. Chronic granulomatous disease. Clin. Rev. Allergy Immunol. 38:3–10.
Huang A., Abbasakoor F., and Vaizey C.J. 2006. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis. 8:637–644.
Kuhns D.B., Alvord W.G., Heller T., Feld J.J., Pike K.M., Marciano B.E., Uzel G., Deravin S.S., Priel D.A., Soule B.P., Zarember K.A., Malech H.L., Holland S.M., and Gallin J.I. 2010. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 363:2600–2610.
Levine S., Smith V.V., Malone M., and Sebire N.J. 2005. Histopathological features of chronic granulomatous disease (CGD) in childhood. Histopathology. 47:508–516.
Lublin M., Bartlett D.L., Danforth D.N., Kauffman H., Gallin J.I., Malech H.L., Shawker T., Choyke P., Kleiner D.E., Schwartzentruber D.J., Chang R., Decarlo E.S., and Holland S.M. 2002. Hepatic abscess in patients with chronic granulomatous disease. Ann. Surg. 235:383–391.
Marciano B.E., Rosenzweig S.D., Kleiner D.E., Anderson V.L., Darnell D.N., Anaya-O’Brien S., Hilligoss D.M., Malech H.L., Gallin J.I., and Holland S.M. 2004. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 114:462–468.
Marciano B.E., Zerbe C.S., Falcone E.L., Ding L., Deravin S.S., Daub J., Kreuzburg S., Yockey L., Hunsberger S., Foruraghi L., Barnhart L.A., Matharu K., Anderson V., Darnell D.N., Frein C., Fink D.L., Lau K.P., Long Priel D.A., Gallin J.I., Malech H.L., Uzel G., Freeman A.F., Kuhns D.B., Rosenzweig S.D., and Holland S.M. 2018. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 141:365–371.
Rieber N., Hector A., Kuijpers T., Roos D., and Hartl D. 2012. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012:252460.
Roos D. and de Boer M. 2014. Molecular diagnosis of chronic granulomatous disease. Clin. Exp. Immunol. 175:139–149.
Roos D., van Bruggen R., and Meischl C. 2003. Oxidative killing of microbes by neutrophils. Microbes Infect. 5:1307–1315.
Schappi M.G., Jaquet V., Belli D.C., and Krause K.H. 2008. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin. Immunopathol. 30:255–271.
Szekely A., Peter M. Jr., Erdos M., and Marodi L. 2012. Hepatic abscess as the single manifestation of X-linked chronic granulomatous disease. Pediatr. Blood Cancer. 58:828–829.
Thomsen I.P., Smith M.A., Holland S.M., and Creech C.B. 2016. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J. Allergy Clin. Immunol. Pract. 4:1082–1088.
Towbin A.J. and Chaves I. 2010. Chronic granulomatous disease. Pediatr. Radiol. 40:657–668; quiz 792–793.
van den Berg J.M., van Koppen E., Ahlin A., Belohradsky B.H., Bernatowska E., Corbeel L., Espanol T., Fischer A., Kurenko-Deptuch M., Mouy R., Petropoulou T., Roesler J., Seger R., Stasia M.J., Valerius N.H., Weening R.S., Wolach B., Roos D., and Kuijpers T.W. 2009. Chronic granulomatous disease: The European experience. PLoS ONE. 4:e5234.
Winkelstein J.A., Marino M.C., Johnston R.B. Jr., Boyle J., Curnutte J., Gallin J.I., Malech H.L., Holland S.M., Ochs H., Quie P., Buckley R.H., Foster C.B., Chanock S.J., and Dickler H. 2000. Chronic granulomatous disease: Report on a national registry of 368 patients. Medicine. 79:155–169.
Zimmermann, A. 2016. Hepatobiliary manifestations of chronic granulomatous diseases of childhood. In Tumors and tumor-like lesions of the hepatobiliary tract. Edited by A. Zimmermann. Cham, Switzerland: Springer. pp. 1–7.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 3 • September 2019
Pages: 107 - 112
History
Received: 2 July 2019
Accepted: 7 August 2019
Accepted manuscript online: 20 August 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
PariaKashani and HaiyingChen. 2019. Histopathological features of patients with chronic granulomatous disease. LymphoSign Journal.
6(3): 107-112. https://doi.org/10.14785/lymphosign-2019-0009
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
