Novel heterozygous NFKB1 mutation in a pediatric patient with cytopenias, splenomegaly, and lymphadenopathy
Abstract
Background: The nuclear factor κ-light-chain enhancer of activated B cells (NF-κB) signaling pathway is a critical regulator of many important adaptive and innate immune responses. The NF-κB transcription factor family consists of 5 structurally related core proteins, one of which is NFKB1. Mutations in the NFKB1 gene has been reported in patients with common variable immune deficiency (CVID) as well as with a large spectrum of clinical features including recurrent viral, bacterial, and fungal infections, autoimmunity, inflammation, and malignancy.
Aim: We describe the clinical characteristics of a pediatric patient with a novel mutation in NFKB1.
Methods: Patient informed consent was obtained in accordance with approved protocols from the Research Ethics Board at the Hospital for Sick Children. Gene panel testing was employed to identify the immune aberration.
Results: Our patient, a previously well 18-month-old boy of Philippines descent, presented with multi-lineage cytopenias (thrombocytopenia, hemolytic anemia, neutropenia), splenomegaly, and lymphadenopathy. He did not have prior history of recurrent infections. Immunological work-up showed normal numbers of T and B cells, normal quantitative immunoglobulins, and adequate vaccination titres. Gene panel testing revealed a novel heterozygous missense variant c.425T>C (p. Ile142Thr) in the NFKB1 gene. Due to persistent cytopenias unresponsive to steroids and IVIG, he was started on Sirolimus with improvement in symptoms.
Conclusion: NFKB1 encodes for p105, which is processed to generate the active p50 transcription factor that can interact with different proteins to activate or inhibit downstream signaling. Our patient was found to have a missense mutation in the Rel homology domain (RHD) of p50, which has distinct functions including DNA binding, protein dimerization, and inhibitory protein binding. The clinical presentation described here broadens the scope of characteristics associated with heterozygous NFKB1 mutations.
Statement of novelty: We report a novel heterozygous missense variant c.425T>C (p. Ile142Thr) in the NFKB1 gene in a pediatric patient with cytopenias, lymphadenopathy, and splenomegaly. To the best of our knowledge, this variant has not been previously reported.
Introduction
The nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling pathway is a critical regulator of many important cellular processes including the innate and adaptive immune responses, cell proliferation and apoptosis, inflammation, and ectodermal development (Karin and Lin 2002; Vallabhapurapu and Karin 2009). Defects in the NF-κB pathway are associated with a large spectrum of human disease presentations manifesting as increased infections, autoimmunity, inflammation, ectodermal dysplasia, and malignancy (Zhang et al. 2017; Scott and Roifman 2019).
The NF-κB transcription factor family consists of 5 structurally related core proteins, NFKB1 (the mature p50 and its precursor p105), NFKB2 (the mature p52 and its precursor p100), RelA, RelB, and c-Rel, which can bind with each other and dimerize to drive or inhibit gene expression at a number of different sites (Siebenlist et al. 1994; Gilmore 2006). Each protein has a conserved N-terminal Rel homology domain (RHD) that allows NF-κB proteins to interact with other proteins (Zhang et al. 2017). In unstimulated cells, NF-κB largely remains in the cytoplasm because of a set of inhibitor proteins comprising the inhibitor of κB (IκB) family (Hayden and Ghosh 2008).
There are 2 well characterized NF-κB signaling pathways, the canonical and non-canonical pathways. The canonical NF-κB pathway is triggered by numerous stimuli including microbial pattern recognition receptors, inflammatory cytokine receptors, or antigen receptors, and leads to phosphorylation of IκBα by the inhibitor of κB kinase (IKK) complex which includes IKKα, IKKβ, and the regulatory protein NF-κB essential modulator (NEMO). Phosphorylation leads to the degradation of IκBα and the subsequent release and translocation of NF-κB into the nucleus (Zhang et al. 2017). The non-canonical NF-κB pathway is activated through binding of a more limited number of receptors including the B-cell activating factor (BAFF) receptor, lymphotoxin β receptor, or CD40 receptor (Shih et al. 2011). Activation leads of this pathway leads to phosphorylation and degradation of different IκB proteins and release of NF-κB into the nucleus. The 2 cascades are likely closely interconnected (Oeckinghaus et al. 2011; Scott and Roifman 2019).
The NFKB1 gene encodes for p105, which is processed to generate the active p50 transcription factor, and can dimerize with different proteins leading to different functions. It can heterodimerize with RelA or c-Rel to activate canonical NF-κB signaling, or form homodimers to inhibit pro-inflammatory gene expression (Kaustio et al. 2017). In recent years, loss of function heterozygous mutations in NFKB1 have been increasingly reported in patients with a previous diagnosis of common variable immunodeficiency (CVID) and others presenting with a wide spectrum of clinical features including infections, auto-inflammation, and malignancy.
We report a novel mutation of NFKB1 in a pediatric patient with cytopenias, lymphadenopathy, and splenomegaly. To the best of our knowledge, this variant has not been reported in previously described NFKB1 cases (Boztug et al. 2016; Maffucci et al. 2016; Schipp et al. 2016; Kaustio et al. 2017; Lougaris et al. 2017; Dieli-Crimi et al. 2018; Fliegauf and Grimbacher 2018; Tuijnenburg et al. 2018).
Methods
Informed consent was obtained in accordance with requirements of the Research Ethics Board at Hospital for Sick Children, Toronto, ON, Canada. Data was gathered both prospectively and retrospectively from available medical records. Genetic testing was performed through Blueprint Genetics, a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory (#99D2092375). The Primary Immunodeficiency Plus Panel (test code IM0301), a 274 gene panel assessing non-coding variants as well as deletion/duplication analysis, was used.
Case presentation
Patient
Our patient, an 18-month-old male, initially presented to a community hospital with petechiae and bruising on his flank, and was found to have thrombocytopenia, DAT+ hemolytic anemia, and neutropenia. He had a history of a respiratory illness 2 weeks prior to presentation. The patient was born at term by spontaneous vaginal delivery to an uncomplicated pregnancy. There were no maternal illnesses, and serology and ultrasounds were normal. He had normal birth length and weight parameters with no dysmorphic features.
The patient was transferred to the Hospital for Sick Children, Toronto, ON, Canada, for further investigations. Following a marrow bone biopsy which was normal (80% cellularity), he started treatment with intravenous immunoglobulin (IVIG) and steroids, with minimal response in his pancytopenia. Abdominal imaging including an ultrasound and magnetic resonance imaging (MRI) demonstrated splenomegaly and intra-abdominal lymphadenopathy. He continued to have ongoing pancytopenia with pronounced thrombocytopenia, and was started on sirolimus (mammalian target of rapamycin [mTOR] inhibitor) 3 months following initial presentation. Following the start of sirolimus, he had improvement in his cytopenias, splenomegaly, and lymphadenopathy. He remained on sirolimus for about 6 months, at which point he was weaned off but unfortunately re-developed thrombocytopenia and neutropenia, and required restarting of sirolimus.
The patient did not have a history of recurrent bacterial, viral, fungal, or opportunistic infections. He did not have any significant atopic disease or skin or nail abnormalities, and no arthritis, mouth ulcers, diarrhea, or change in stool habits. He did not have any constitutional symptoms such as fever, weight loss, or decreased energy. His vaccinations were up to date and he was developmentally appropriate for his age. Family history was significant for systemic lupus erythematosus in his paternal grandmother, and 2 paternal grandaunts who both died of renal failure. He had an older sister who was healthy. His parents were of Philippines descent with no known consanguinity.
On physical examination, there were no dysmorphic features. His skin exam was significant for mild bruising and petechiae on his right abdomen. He did not have any palpable lymphadenopathy or evidence of organomegaly. The remainder of his cardiovascular, respiratory, musculoskeletal, and neurological exams were unremarkable.
General investigations
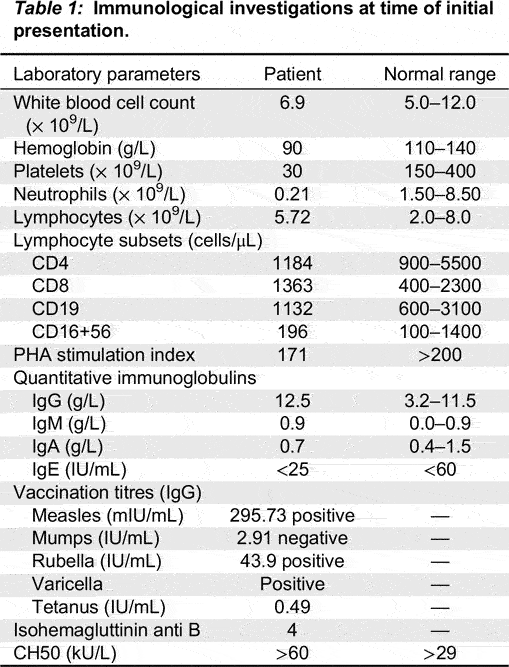
At initial presentation, the patient’s complete blood count demonstrated total white blood cell count of 6.9 × 109/L, hemoglobin 90 g/L, platelet count 30 × 109/L, neutrophils 0.21 × 109/L, and lymphocytes 5.72 × 109/L. DAT was positive with reticulocytosis. Electrolytes, renal function, and liver enzymes were normal. He had a normal vitamin B12 level and normal numbers of peripheral double negative T cells. Extensive infectious disease investigations showed negative testing for hepatitis A, B, C, HIV, EBV, CMV, HSV, and HHV6. Metabolic work-up for an underlying metabolic disorder was unremarkable.
Immunological investigations
The patient’s immunological investigations are summarized in Table 1. Humoral evaluation demonstrated elevated IgG of 12 g/L with normal IgA, IgM, and IgE. Vaccination titres demonstrated positive reactivity to measles, rubella, and tetanus and negative to mumps. T cell evaluation showed normal numbers of T, B, and NK cells with low T cell proliferation, however PHA test was performed while patient was on sirolimus, and shortly after being on IVIG. The patient has not been off of sirolimus for a long enough period for repeat of the PHA to be repeated.
Table 1:
Genetic testing
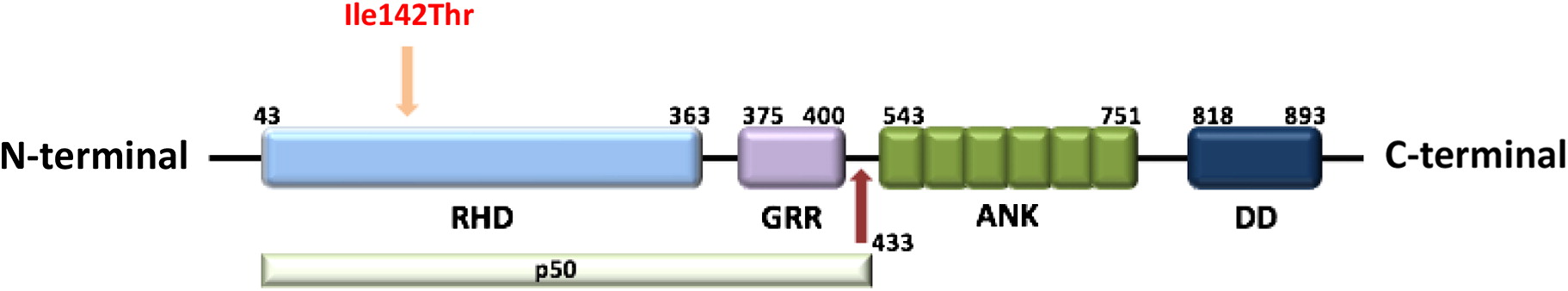
The patient’s clinical features of multi-lineage cytopenias, lymphadenopathy, and splenomegaly was concerning for an underlying immunodeficiency or immune dysregulation disorder. Initial targeted Sanger gene sequencing of FAS and PIK3CD genes were normal. Panel genetic testing was then conducted utilizing a commercially available Primary Immunodeficiency panel. He was found to have a novel heterozygous missense variant in the NFKB1 gene (NM_003998.3), c.425T>C, resulting in p. Ile142Thr (Figure 1). There is a strong association between the gene and this patient’s phenotype, and variant is predicted to be damaging by in silico tools. This genetic variant has not been observed in the reference population cohorts of the Genome Aggregation Database (gnomAD, n > 120 000 exomes and >15 000 genomes), nor reported in disease-related variation databases such as ClinVar or the Human Gene Mutation Database (HGMD).
Figure 1:
Discussion
We describe a young pediatric patient who first presented with severe thrombocytopenia, neutropenia, splenomegaly, and lymphadenopathy at the age of 18 months. He was found to have a novel missense mutation in the NFKB1 gene that lead to a change from isoleucine to threonine at the 142 amino acid position in the Rel homology domain (RHD) of p50. This variant has not yet been reported in the literature (Table 2) and likely explains his clinical presentation and has possible future implications for the development of other symptoms in the future.
Table 2:

The RHD is composed of 300 amino acids and has 3 distinct functions including DNA binding, protein dimerization, and inhibitory protein binding (Hayden et al. 2012). Mutations in the RHD has been reported in a number of families with variable clinical phenotype. In a Dutch-Australian family with CVID, a splice-site mutation was found to cause in-frame skipping of exon 8 which lead to degradation of the altered protein (Fliegauf and Grimbacher 2018). Similarly, 2 other families with heterozygous frameshift mutations in RHD were found to have decreased expression of p50. Haploinsufficiency of NFKB1 in these 3 families presented with a heterogeneous spectrum of illness including hypogammaglobulinemia, recurrent sinopulmonary infections, autoimmunity, skin lesions, colitis, lymphoproliferation, and malignancy (Fliegauf and Grimbacher 2018). Interestingly, 1 of the patients in the New Zealand family with a heterozygous frameshift mutation (c.465dupA) in exon 7, presented with similar symptoms as our patient, consisting of thrombocytopenia, hemolytic anemia, and neutropenia from the young age of 2 years in the absence of other symptoms (Fliegauf and Grimbacher 2018).
Multiple recent studies have identified variants in NFKB1 as a monogenic cause of immunodeficiency/immune dysregulation (Table 2) (Maffucci et al. 2016; Schipp et al. 2016; Kaustio et al. 2017; Lougaris et al. 2017; Dieli-Crimi et al. 2018; Fliegauf and Grimbacher 2018; Tuijnenburg et al. 2018). Patients display a wide range of phenotypic heterogeneity including recurrent sinopulmonary infections, viral and fungal infections, autoimmunity, lymphoproliferation, and malignancy. Differences in gene dosage, modifier genes, and post-transcriptional elements may account for the wide spectrum of penetrance and expressivity.
Heterozygous mutations in 3 Finnish families have been reported in patients with antibody deficiency, recurrent infections, and auto-inflammation (arthritis, mouth ulcers, hyper-inflammatory response to surgery) (Kaustio et al. 2017). Two families had mutations in the RHD of p50 and 1 had a mutation in the Ankyrin repeats of p105. The NFKB1 variants were associated with different changes in downstream signaling including reduction in p50 and p105 protein quantity, enhanced degradation of p105, and reduced nuclear entry of p50 and decreased transcriptional activity (Kaustio et al. 2017). There was also increased secretion of pro-inflammatory cytokines interleukin-1-β (IL-1β) and tumor necrosis factor (TNF) in 1 of the kindreds (Kaustio et al. 2017). All patients developed symptoms in adulthood and no patients had a similar presentation to our patient from such a young age.
In a cohort of 846 unrelated patients with primary immunodeficiency, including 390 patients with CVID, 16 patients were identified to have pathogenic mutations in NFKB1 (Tuijnenburg et al. 2018). All 16 patients were within the CVID subgroup, and mutations in NFKB1 accounted for 4% of all patients with CVID, which was the most common monogenic defect in this group of patients with CVID. Individual NFKB1 variants were classified as high-effect variants (large deletion, nonsense, frameshift, and splice site variants) or moderate effect (missense substitutions). Interestingly, a large number of missense NFKB1 variants were identified (52), the majority of which were predicted to not be associated with pathogenicity. Three variants were unique in the cohort of patients with CVID and classified as likely pathogenic. Consistent with other reports of patients with mutations in NFKB1, there was a high degree of variability in both the penetrance and expressivity associated with NFKB1 mutations (Tuijnenburg et al. 2018).
Although haploinsufficiency of NFKB1 has been predominantly described as a defect of B cells, there are reported cases of diminished T cell function, EBV-associated lymphoproliferation, and viral and fungal infections, suggesting that it may also affect T cell function (Boztug et al. 2016; Schipp et al. 2016). One patient first presented with autoimmune hemolytic anemia at 14 years of age, then developed lymphadenopathy, hepatosplenomegaly, and frequent viral, bacterial, and fungal infections (Schipp et al. 2016). She had hypogammaglobulinemia, decreased class-switched B cells, naïve CD4 and regulator T cells, and increased double negative T cells. She was found to have a frameshift mutation in RHD which lead to decreased protein expression (Schipp et al. 2016). In our patient, T cell numbers were normal but the T cell proliferation response was low, although it was performed while the patient was on sirolimus and shortly after IVIG treatment. This will need to be repeated in the future, however at this time, he does not have evidence of any severe viral, fungal, or opportunistic infections suggestion of aberrant T cell function.
NFKB1 knockout mice have been shown to develop multi-organ autoimmunity, as well as aberrant immune responses involving B cell dysfunction and abnormal maturation of T regulatory cells. Increased production of pro-inflammatory cytokines including interferon-γ (IFNγ) and tumor necrosis factor (TNF) has also been reported. These mice have increased susceptibility to infection and gut inflammation, which has also been seen in patients with NFKB1 haploinsufficiency (Stanic et al. 2004; de Valle et al. 2016; Schipp et al. 2016; Kaustio et al. 2017; Dieli-Crimi et al. 2018). To further delineate the biological implications of this novel mutation in our patient, additional functional studies are warranted to ascertain the degree of p50 protein expression, activation of downstream signaling elements such as phosphorylation, and production of cytokines.
Our patient was started on sirolimus for his cytopenias, lymphadenopathy, and splenomegaly. He had improvement in his symptoms following the start of treatment and recurrence when it was discontinued. Sirolimus is an mTOR inhibitor which inhibits cytokine receptor-dependent signal transduction, blocking the activation of T and B lymphocytes (Sehgal 2003). It can be taken orally, is safe, and has been used in a prospective trial of patients with refractory multi-lineage cytopenias from different underlying diagnoses including autoimmune lymphoproliferative syndrome (ALPS), CVID, systemic lupus erythematosus (SLE), and single lineage cytopenias (Bride et al. 2015). Current treatment options for patients with NFKB1 mutations and CVID clinical presentation including symptomatic treatment with immunoglobulin replacement, immunosuppression, and (or) antibiotics. Better delineation of the pathogenic mechanisms associated with NFKB1 variants may inform more targeted treatment strategies.
Conclusion
We report a novel mutation in the RHD domain of NFKB1 in a pediatric patient with early onset cytopenias, splenomegaly, and lymphadenopathy. This case demonstrates the importance of considering primary immunodeficiency in patients with unexplained autoimmune cytopenias, and further contributes to the growing clinical phenotype of heterozygous NFKB1 mutations.
REFERENCES
Boztug H., Hirschmugl T., Holter W., Lakatos K., Kager L., Trapin D., Pickl W., Förster-Waldl E., and Boztug K. 2016. NF-κB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J. Clin. Immunol. 36:533–540.
de Valle E., Grigoriadis G., O’Reilly L.A., Willis S.N., Maxwell M.J., Corcoran L.M., Tsantikos E., Cornish J.K.S., Fairfax K.A., Vasanthakumar A., Febbraio M.A., Hibbs M.L., Pellegrini M., Banerjee A., Hodgkin P.D., Kallies A., Mackay F., Strasser A., Gerondakis S., and Gugasyan R. 2016. NFκB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J. Exp. Med. 213:621–641.
Dieli-Crimi R., Martínez-Gallo M., Franco-Jarava C., Antolin M., Blasco L., Paramonov I., Semidey M.E., Fernández A.Á., Molero X., Velásquez J., Martín-Nalda A., Pujol-Borrell R., and Colobran R. 2018. Th1-skewed profile and excessive production of proinflammatory cytokines in a NFKB1-deficient patient with CVID and severe gastrointestinal manifestations. Clin. Immunol. 195:49–58.
Fliegauf M. and Grimbacher B. 2018. Nuclear factor κB mutations in human subjects: The devil is in the details. J. Allergy Clin. Immunol. 142:1062–1065.
Gilmore T.D. 2006. Introduction to NF-κB: Players, pathways, perspectives. Oncogene. 25:6680–6684.
Hayden M.S. and Ghosh S. 2008. Shared principles in NF-κB signaling. Cell. 132:344–362.
Hayden M.S. and Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26:203–234.
Karin M. and Lin A. 2002. NF-κB at the crossroads of life and death. Nat. Immunol. 3:221–227.
Kaustio M., Haapaniemi E., Göös H., Hautala T., Park G., Syrjänen J., Einarsdottir E., Sahu B., Kilpinen S., Rounioja S., Fogarty C.L., Glumoff V., Kulmala P., Katayama S., Tamene F., Trotta L., Morgunova E., Krjutškov K., Nurmi K., Eklund K., Lagerstedt A., Helminen M., Martelius T., Mustjoki S., Taipale J., Saarela J., Kere J., Varjosalo M., and Seppänen M. 2017. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J. Allergy Clin. Immunol. 140:782–796.
Lougaris V., Moratto D., Baronio M., Tampella G., Meer J.W.M., van der Badolato R., Fliegauf M., and Plebani A. 2017. Early and late B-cell developmental impairment in nuclear factor kappa B, subunit 1-mutated common variable immunodeficiency disease. J. Allergy Clin. Immunol. 139:349–352.e1.
Maffucci P., Filion C.A., Boisson B., Itan Y., Shang L., Casanova J.-L., and Cunningham-Rundles C. 2016. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front. Immunol. 7:220.
Oeckinghaus A., Hayden M.S., and Ghosh S. 2011. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12:695–708.
Schipp C., Nabhani S., Bienemann K., Simanovsky N., Kfir-Erenfeld S., Assayag-Asherie N., Oommen P.T., Revel-Vilk S., Hönscheid A., Gombert M., Ginzel S., Schäfer D., Laws H.-J., Yefenof E., Fleckenstein B., Borkhardt A., Stepensky P., and Fischer U. 2016. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica. 101:e392–e396.
Scott O. and Roifman C.M. 2019. NF-κB pathway and the Goldilocks principle: Lessons from human disorders of immunity and inflammation. J. Allergy Clin. Immunol. 143:1688–1701.
Sehgal S.N. 2003. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 35:S7–S14.
Shih V.F.-S., Tsui R., Caldwell A., and Hoffmann A. 2011. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 21:86–102.
Siebenlist U., Franzoso G., and Brown K. 1994. Structure, regulation and function of NF-κB. Annu. Rev. Cell Biol. 10:405–455.
Stanic A.K., Bezbradica J.S., Park J.-J., Matsuki N., Mora A.L., Van Kaer L., Boothby M.R., and Joyce S. 2004. NF-κB controls cell fate specification, survival, and molecular differentiation of immunoregulatory natural T lymphocytes. J. Immunol. 172:2265–2273.
Tuijnenburg P., Lango Allen H., Burns S.O., Greene D., Jansen M.H., Staples E., Stephens J., Carss K.J., Biasci D., Baxendale H., Thomas M., Chandra A., Kiani-Alikhan S., Longhurst H.J., Seneviratne S.L., Oksenhendler E., Simeoni I., de Bree G.J., Tool A.T.J., van Leeuwen E.M.M., Ebberink E.H.T.M., Meijer A.B., Tuna S., Whitehorn D., Brown M., Turro E., Thrasher A.J., Smith K.G.C., Thaventhiran J.E., Kuijpers T.W., and NIHR BioResource—Rare Diseases Consortium. 2018. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 142:1285–1296.
Vallabhapurapu S. and Karin M. 2009. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27:693–733.
Zhang Q., Lenardo M.J., and Baltimore D. 2017. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell. 168:37–57.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 2 • June 2019
Pages: 61 - 67
History
Received: 8 April 2019
Accepted: 22 May 2019
Accepted manuscript online: 24 May 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
LucyDuan and StephenFeanny. 2019. Novel heterozygous NFKB1 mutation in a pediatric patient with cytopenias, splenomegaly, and lymphadenopathy. LymphoSign Journal.
6(2): 61-67. https://doi.org/10.14785/lymphosign-2019-0006
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
