Successful hematopoietic stem cell transplantation in a patient with a novel mutation in coronin 1A
Abstract
Introduction: Coronin 1A is part of a family of highly conserved actin regulatory proteins with key roles in T cell homeostasis and T cell receptor signaling. Null mutations in coronin 1A result in severe combined immunodeficiency, whereas hypomorphic mutations have been associated with a somewhat milder immunological phenotype. Nevertheless, all patients described so far have markedly reduced naïve peripheral T cells, impaired T cell responses to mitogens, and limited T cell receptor diversity. Interestingly, despite poor thymic output, thymus architecture appears normal. To date, only 2 cases of hematopoietic stem cell transplantation (HSCT) have been reported in coronin 1A deficiency.
Aim: To describe the identification, transplantation course, and long term outcome of a Canadian Inuit patient diagnosed with coronin 1A deficiency.
Methods: Patient chart review was performed in accordance with institutional research ethics approval. A combination of immunological investigations and molecular genetic analyses were utilized to identify a novel mutation in the tryptophan-aspartate repeat region of coronin 1A. Based on the patient’s profound T cell dysfunction, the decision was made to proceed with HSCT.
Results: The patient presented with a history of recurrent urinary tract infections, otitis media, and developmental delay involving poor axial and peripheral muscle tone. Axillary lymphadenopathy was noted and subsequent thymus biopsy revealed aberrant CD7+ T cell deficiency. Lymphocyte responses to mitogens and T cell receptor excision circle levels were markedly reduced, consistent with the diagnosis of severe combined immunodeficiency. Whole exome sequencing and Sanger confirmation revealed a novel mutation in coronin 1A. HSCT using a HLA-matched unrelated donor resulted in long term engraftment and solid immune reconstitution.
Conclusion: Very few patients with coronin 1A deficiency have been described to date, making it difficult to evaluate its natural history and management. Here, we describe the presentation, identification, transplantation, and outcome in our patient.
Statement of novelty: We describe the successful hematopoietic stem cell transplantation course and outcome in a patient with a novel mutation in coronin 1A.
Introduction
Coronin 1A is a key regulatory protein involved in cytoskeletal remodeling and calcium signaling (Rybakin and Clemen 2005; Mueller et al. 2008), and is expressed in a number of hematopoietic cells, including lymphocytes, macrophages, and NK cells (Oku et al. 2003). Part of a larger family of highly conserved actin regulatory proteins (Xavier et al. 2008), the structure of coronin 1A comprises: (i) an amino tryptophan-aspartate (WD) repeat-containing region forming a beta propeller—a motif involved in plasma membrane binding; (ii) a unique region; and (iii) a leucine zipper coiled-coil domain that is required for oligomerization and cytoskeletal association (Gatfield et al. 2005; Kammerer et al. 2005).
The 46 amino acid, 57 kDa actin binding protein is encoded by the CORO1A gene, located on chromosome 16p11.2 (Suzuki et al. 1995). Essential roles for coronin 1A have been reported in cell survival, migration, phagocytosis, vesicular trafficking, and signal transduction (Rybakin and Clemen 2005; Foger et al. 2006; Mueller et al. 2008, 2011). In murine studies, loss of coronin 1A expression is associated with T lymphocytopenia, due in part to aberrant T cell receptor mediated proliferation and failure of T cells to progress through the cell cycle (Foger et al. 2006; Haraldsson et al. 2008; Mueller et al. 2008; Mugnier et al. 2008; Shiow et al. 2008). Dysfunctional CD4+ T helper cell and associated IgG responses have also been reported (Tchang et al. 2013), while B and NK cells remain at normal levels. In humans, null mutations in CORO1A resulting in complete absence of coronin 1A expression are associated with a severe combined immunodeficiency phenotype (Shiow et al. 2008), while hypomorphic mutations lead to somewhat milder immunological manifestations (Moshous et al. 2013; Yee et al. 2016). In all cases described so far, patients experienced recurrent infections, had markedly reduced naïve peripheral T cells, and impaired T cell responses to mitogens (Moshous et al. 2013; Mace and Orange 2014). All but 3 patients showed failure to control Epstein-Barr virus (EBV) predisposing to B cell lymphoproliferation and lymphoma (Yee et al. 2016; Dinur Schejter et al. 2019). Interestingly, the thymus of coronin 1A deficient patients appears normal, indicating that loss of naïve T cells may be due to defects in cell survival. Variability in humoral function, B and NK cell counts, as well as T cell responses to mitogens and antigens have been described among affected individuals.
To date, only 2 patients with coronin 1A deficiency are reported to have received hematopoietic stem cell transplantation (HSCT). The first had a human leukocyte antigen (HLA)-matched unrelated donor transplant at the age of 4 years (Shiow et al. 2009). Although the procedure was successful, a full account of long term outcome has not been provided. The second patient received 6/6 HLA-matched parental bone marrow, however, died at 4 months post-transplant due to respiratory failure (Moshous et al. 2013).
Here, we describe a novel mutation in coronin 1A in a patient of Canadian Inuit origin. The patient successfully underwent HSCT using a HLA-matched unrelated donor and achieved long term engraftment and solid immune reconstitution.
Methods
Patient
Patient chart review was performed following informed consent, and in accordance with a research ethics approved protocols.
Serum concentration of immunoglobulin and specific antibodies
Serum concentrations of immunoglobulins were measured by nephelometry. Levels of serum antibodies to tetanus were measured by ELISA.
T and B cell proliferative response
Lymphocyte proliferative responses to mitogens including phytohemagglutinin (PHA) and anti-CD3. All assays were performed in triplicate and were compared with simultaneously stimulated normal controls.
Exome sequencing and variant calling
DNA from blood was submitted to The Centre for Applied Genomics (TCAG), Toronto, ON, Canada for exome library preparation and sequencing. DNA was quantified by Qubit DNA HS assay (Life Technologies, Carlsbad, CA, USA) and 100 ng of input DNA was used for library preparation using the Ion AmpliSeq Exome Kit (Life Technologies) according to the manufacturer’s recommendations. The Ampliseq Exome library was immobilized on Ion PI™ Ion Sphere™ particles using the Ion PI Template OT2 200 Kit v3. Sequencing was performed with the Ion PI Sequencing 200 Kit v3 and Ion PI Chip v2 in the Ion Proton™ semiconductor sequencing system following the manufacturer’s recommendation.
Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server, using the Ion Proton ampliseq germline low stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline (Stavropoulos et al. 2016) based on Annovar (November 2014 version) (Wang et al. 2010) and RefSeq gene models (downloaded from UCSC 1 August 2015).
Sequencing analysis
Patient’s genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers designed upstream and downstream of the coronin 1A gene. Sequencing was done using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Western blotting
Whole-cell lysates were prepared in a 1% Triton X-100 buffer and analyzed by Western blotting. Anti-coronin 1A antibodies were purchased from Creative Diagnostics Inc. and Giα3 were purchased from Santa Cruz Biotechnology Inc.
Results
Case presentation
The female patient was born at term to a single mother of a 4 year old male, and is a product of consanguineous Canadian Inuit parents. Serology during pregnancy was negative for hepatitis B and HIV. Group B Streptococcus was positive and during labor she received 3 doses of penicillin G. There were no labor or postnatal complications and her birth weight was 3882 g. As per routine practice in her community, she received the live-attenuated BCG vaccination shortly after birth.
At 1 month of age, the patient had vomiting episodes which raised the possibility of pyloric stenosis. At 2 months of age, she suffered a right upper lobe pneumonia and the nasopharyngeal swab was positive for parainfluenza at the time. At 6 months of age, she had recurrent urinary tract infections and urine cultures grew Escherichia coli and Klebsiella. A renal ultrasound was normal.
Developmental assessment at 6 months revealed a delay as she was not sitting up on her own. Her muscle tone was decreased axially and peripherally. At 6 months of age, she was admitted for diarrhea that lasted for 3 weeks. Clostridium difficile was detected in the stool and she was given a 10 day course of metronidazole. She also suffered chronic suppurative otitis media bilaterally since the age of 3 months.
X-ray revealed some atelectasis and chronic changes in the upper right lobe. Blood counts revealed anemia with a hemoglobin level of 104 g/L, a low white cell count of 3.9 cells/μL and low lymphocyte count of 0.5 × 109 cells/L.
At the age of 7 months, she was transferred to a tertiary care center and was noticed to have axillary lymphadenopathy. A lymph node biopsy showed confluent well-formed non-necrotizing granulomas, while architecture and cellularity were normal. Ziehl–Neelsen staining revealed abundant acid-fast bacilli and cultures grew Mycobacterium bovis. Bronchoalveolar lavage fluid microbiological cultures were negative including mycobacteria. She was started on appropriate anti-mycobacterials. Circulating lymphocytes were low with only 135 CD4+ and 15 CD8+ cells/μL. Responses to mitogens were markedly reduced and T cell receptor excision circle (TREC) levels were only 2.
Immune evaluation
At 7 months of age the patient had profound lymphopenia. Flow cytometry revealed 202 CD3+, 135 CD4+ and 34 CD8+ T cells/μL, 194 CD19+ B cells/μL and normal numbers of circulating CD16/56+ NK cells/μL. Most CD3+ and CD4+ T cells were also CD45RO+, suggesting poor thymus output. In vitro responses to PHA and anti-CD3 were borderline reduced (Table 1).
Table 1:
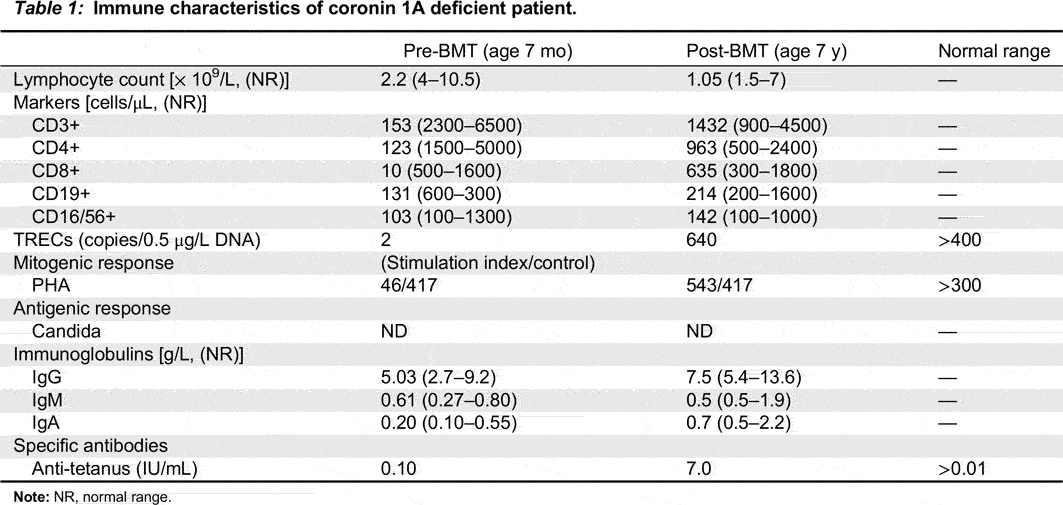
Note: NR, normal range.
In contrast, immunoglobulin levels were normal for age and antibody levels to Tetanus toxoid was protective, suggesting some partial T cell function.
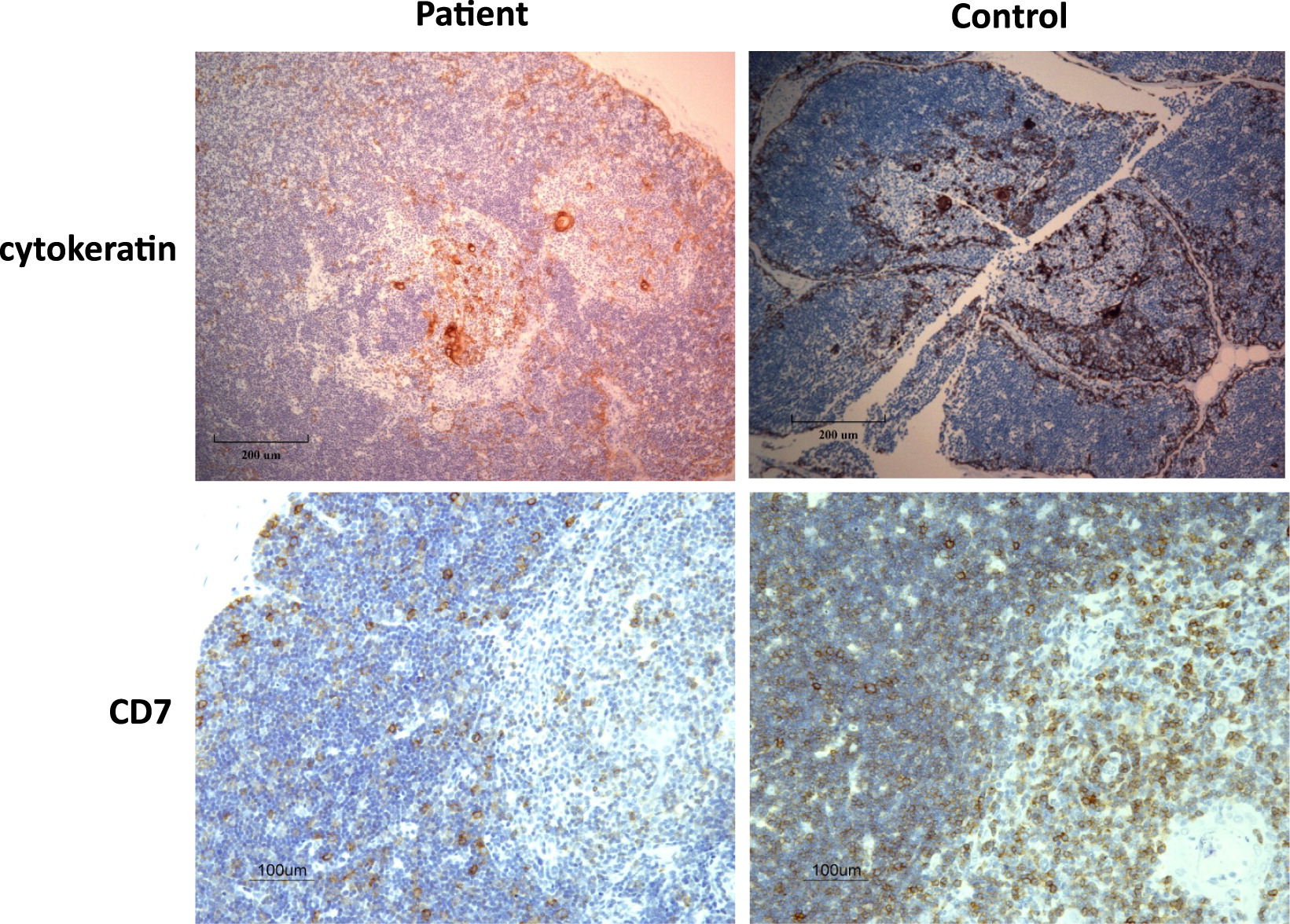
To ascertain the diagnosis, a thymus biopsy was performed which revealed an architecturally normal thymus with medullary Hassall’s corpuscles and normal corticomedullary demarcation. However, an overall aberrant deficiency of CD7+ T cells was noted (Figure 1).
Figure 1:
Analysis of CD4+ T cell repertoire demonstrated reduced representation of Vβ1, Vβ4, Vβ5.1, Vβ5.2, Vβ5.3, Vβ9, and Vβ18, whereas Vβ2, Vβ7.1, and Vβ23.3 were over represented. Similarly, CD8+ T cell repertoire was skewed showing under representation of Vβ1, Vβ4, Vβ5.1, Vβ5.3, Vβ7.1, Vβ8, Vβ9, Vβ11, Vβ13.1, Vβ13.6, Vβ14, Vβ17, Vβ21.3, and Vβ22 (Figure 2).
Figure 2:
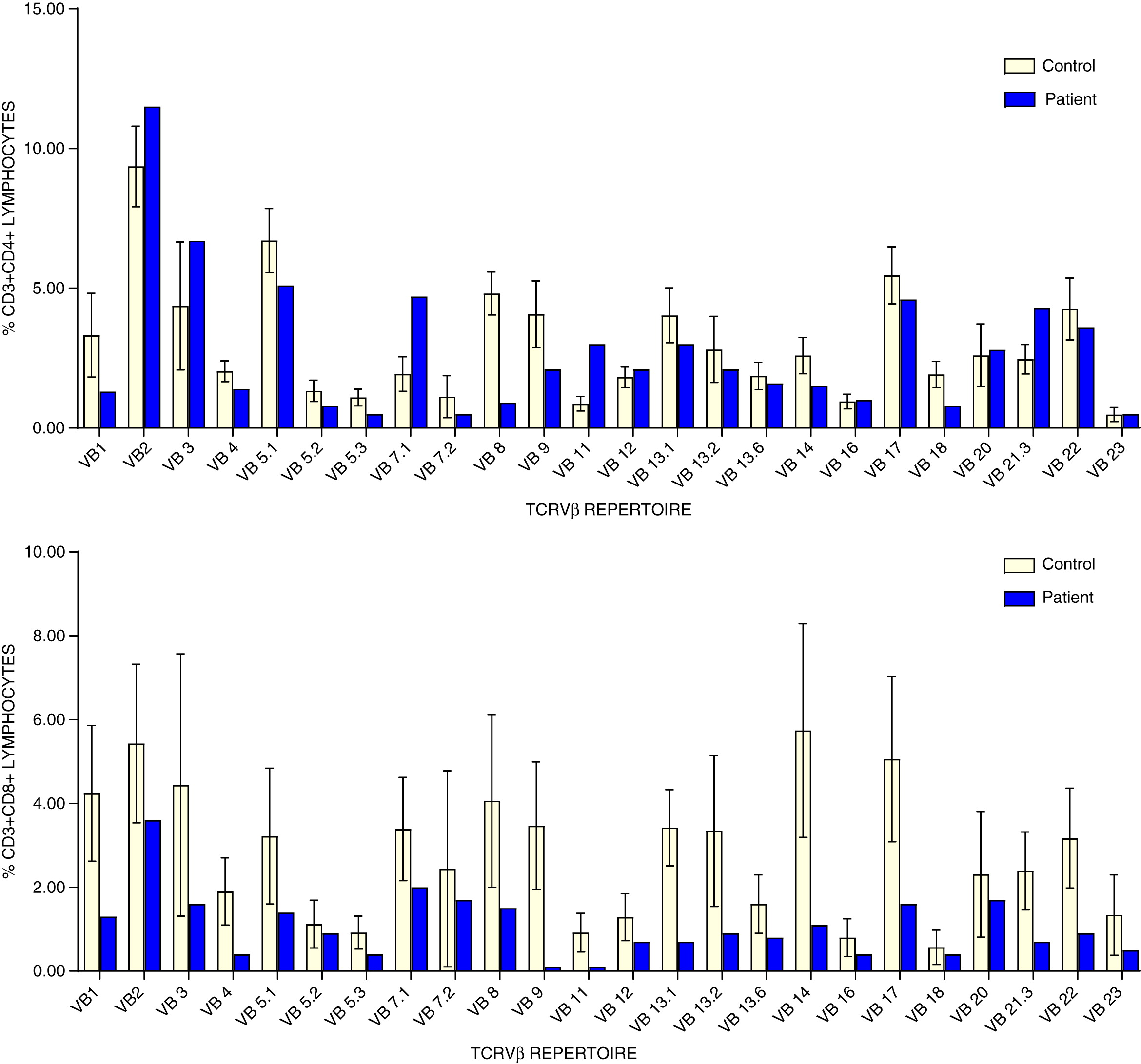
Retrospective measurement of TREC from the patient’s Guthrie dried blood spot revealed just 1 copy/3 μL, a positive screen for SCID.
Genetic analysis
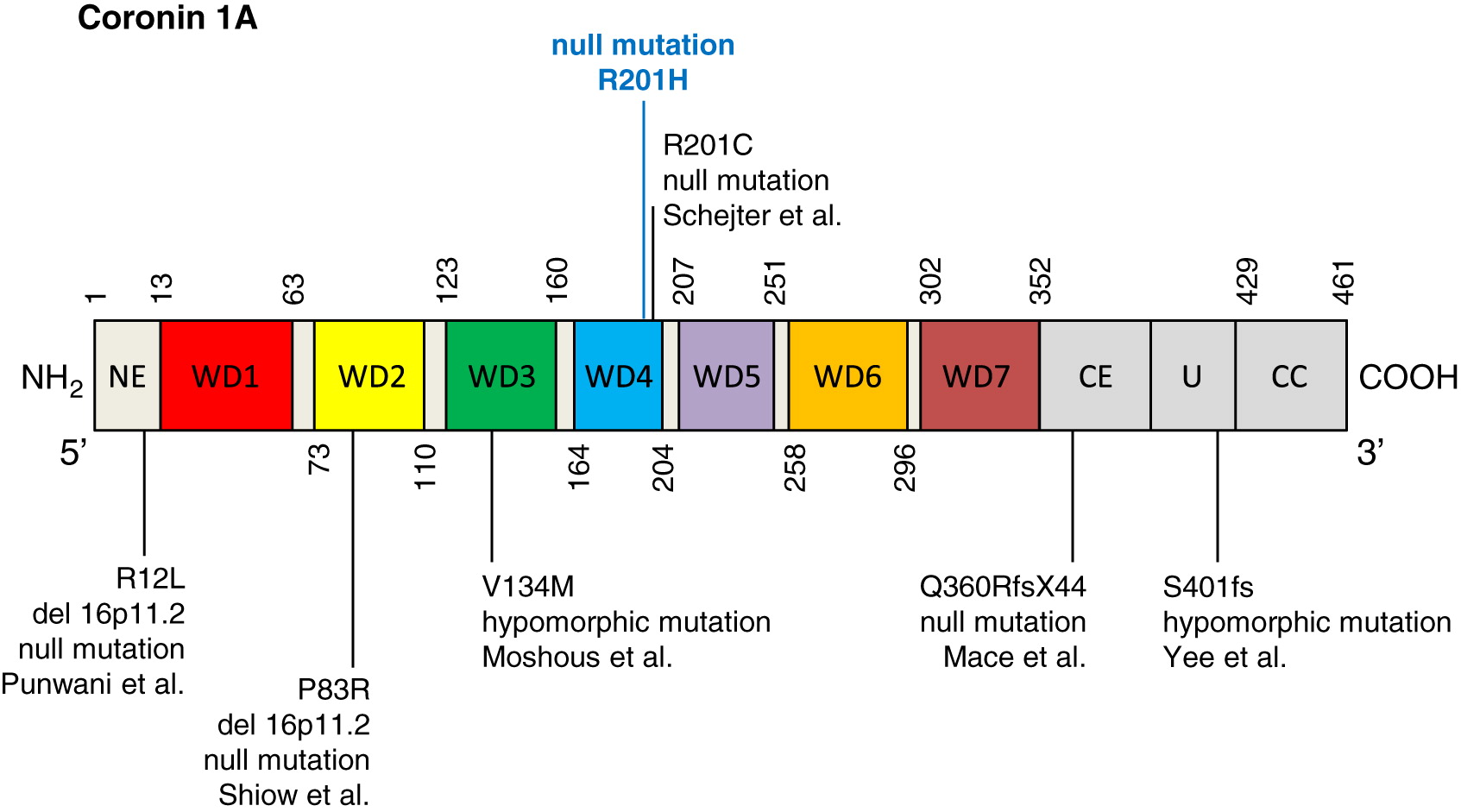
Whole exome sequencing identified a novel homozygous missense mutation in CORO1A, c. 602G>A (p.R201H), and was confirmed by Sanger sequencing (Figure 3). The mutation is localized to the WD repeat-containing region of coronin 1A (Figure 4). The patient’s mother was found to be a heterozygous carrier. Genetic analysis of the patient’s father was not available.
Figure 3:
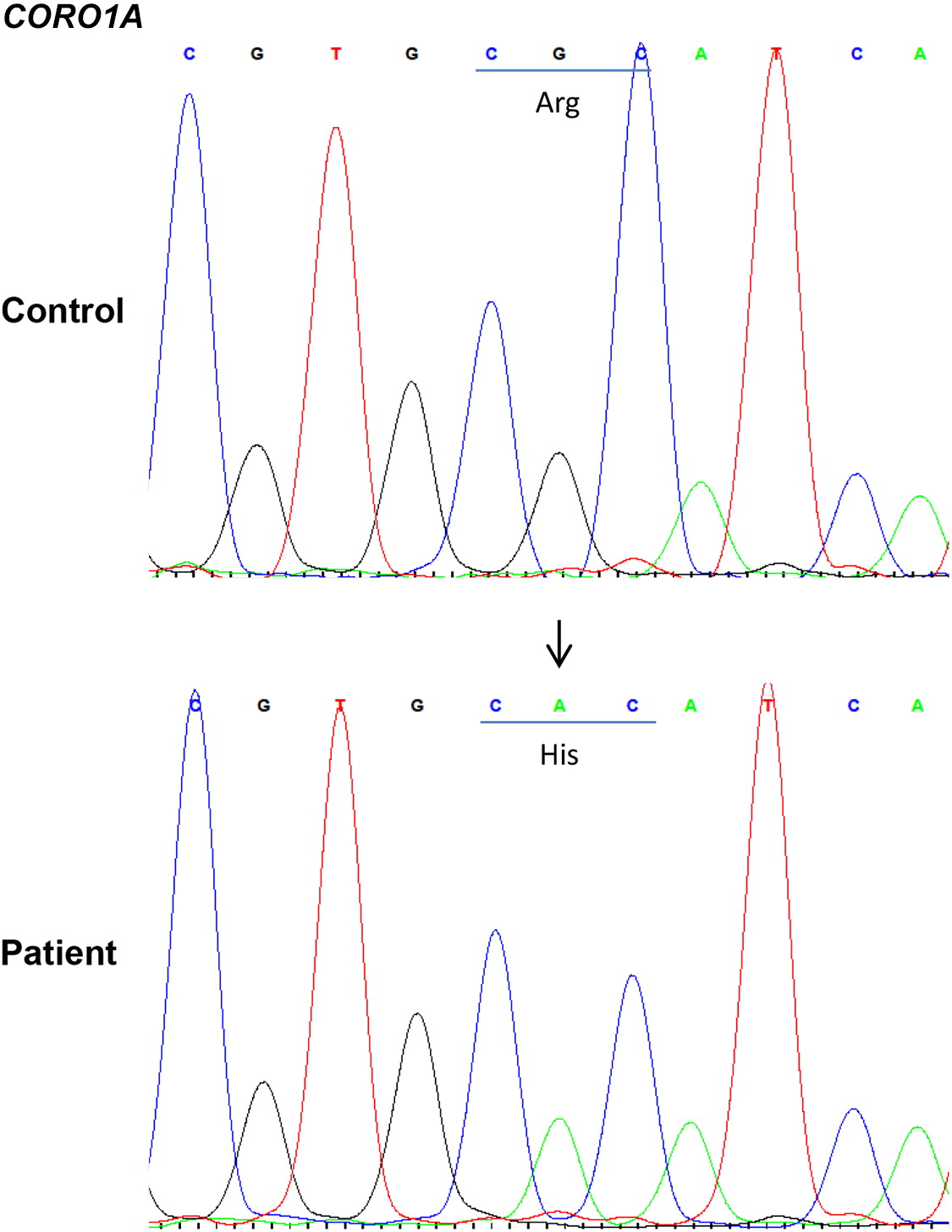
Figure 4:
Coronin 1A protein expression
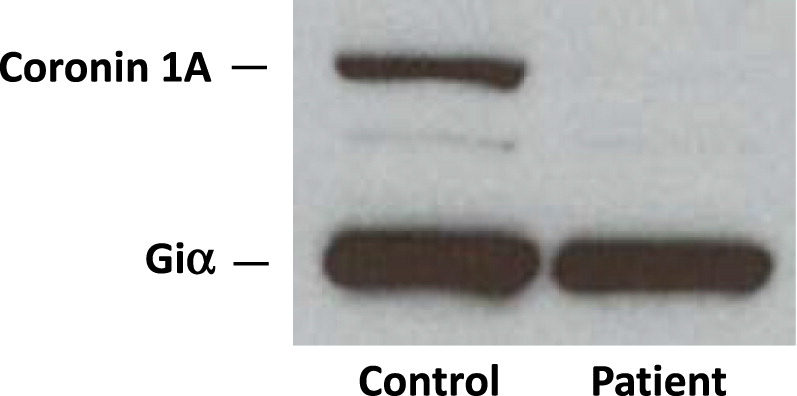
Western blotting confirmed the complete lack of coronin 1A protein expression (Figure 5).
Figure 5:
HSCT course, engraftment, and outcome
At 13 months of age, the patient received a 9/10 HLA-matched unrelated donor transplant after myeloablative conditioning with busulfan, cyclophosphamide, and alemtuzumab. Neutrophil engraftment occurred on day 17 post-transplant. She had 1 infectious episode with Pseudomonas aeruginosa bacteremia at day 4 post-transplant, which responded rapidly to antibiotic treatment. Cyclosporine and methyl prednisone prophylaxis prevented acute graft versus host disease. However, at 1.5 years post-transplant she had an episode of fever associated with generalized rash, hemolytic anemia, and pancytopenia. Bone marrow biopsy showed normal cellularity. She was successfully treated with steroids and cyclosporine, but subsequently developed repeated episodes of hemolytic anemia and partial scalp alopecia which again responded well to immunosuppressive medications.
By 2 years post-transplant her immune evaluation demonstrated full engraftment (86% donor cells) as well as solid immune reconstitution with normal TREC levels and normal responses to mitogens and antigens. She continued anti-mycobacterials (isoniazid, rifampin) throughout the transplant course without evidence of BCG disease. These were discontinued around age 3. She did not develop evidence of graft versus host disease. Currently, at the age of 7 years, her engraftment and immune function remain solid (Table 1).
Discussion
Very few patients with coronin 1A deficiency have been described so far, making it difficult to evaluate its natural history and management. The challenge stems from the observation that the clinical features as well as the degree of immune dysfunction are highly variable in these patients.
Our patient presented with a phenotype of severe combined immunodeficiency; immune evaluation revealed low circulating T lymphocytes, skewed T cell repertoire, markedly reduced lymphocyte proliferative responses to mitogens, and TREC level (an indicator of thymic output) of only 2. The thymus, while architecturally normal, was deficient in CD7+ T cells. In an effort to identify the underlying cause of immunodeficiency, we performed whole exome sequencing and identified a novel homozygous missense mutation in CORO1A (c.602G>A), resulting in p.R201H within the WD repeat containing region.
In cases where profound T cell dysfunction has been documented, it is plausible to consider curative HSCT. We thus proceeded with a 9/10 HLA-matched unrelated donor transplant when the patient was 13 months of age. At 2- and 7-years post-transplant, her immune evaluations revealed full engraftment, robust immune reconstitution, as well as normal TREC levels and T cell responses to mitogens and antigens.
To date, there have been reports of 10 patients (from 6 kindreds) with coronin 1A deficiency (Shiow et al. 2008; Moshous et al. 2013; Stray-Pedersen et al. 2014; Punwani et al. 2015; Yee et al. 2016; Dinur Schejter et al. 2019). Here, we describe an 11th patient with a novel mutation in the WD4 repeat which is necessary for formation of the beta propeller motif. Mutations affecting the N-terminal extension (R12L/del116p11) (Punwani et al. 2015), WD repeat regions—WD2 (P83R/del116p11) (Shiow et al. 2008), WD4 (R201C) (Dinur Schejter et al. 2019), and C-terminal extension (Q360RfsX44) (Stray-Pedersen et al. 2014) have each been associated with complete loss of coronin 1A protein expression, either through truncation codons leading to nonsense mediated decay of mRNA or possibly loss of the stabilizing coiled-coil domain. In cases where residual albeit low levels of coronin 1A were detected (V134M affecting WD3, and S401fs targeting the CE region), a somewhat milder immunological phenotype was described. Nevertheless, attempts to assign genotype–phenotype correlations are complicated by the relatively small number of affected individuals, variability observed in time of disease onset, as well as novel manifestations that continue to broaden the spectrum of disease. With the increased use of next generation sequencing techniques, it is likely that the identification of further coronin 1A deficient patients will help to establish effective evidence-based treatment recommendations.
We have shown here that HSCT using a HLA-matched unrelated bone marrow donor is a curative therapeutic option for coronin 1A deficiency, and results in long term engraftment and solid immune reconstitution. This report also revealed for the first time a novel mutation identified in a patient of Canadian Inuit origin.
REFERENCES
Dinur Schejter Y., Mandola A., and Reid B. 2019. Coronin 1A deficiency identified by newborn screening for severe combined immunodeficiency. LymphoSign J. 6:17–25.
Foger N., Rangell L., Danilenko D.M., and Chan A.C. 2006. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science. 313:839–842.
Gatfield J., Albrecht I., Zanolari B., Steinmetz M.O., and Pieters J. 2005. Association of the leukocyte plasma membrane with the actin cytoskeleton through coiled coil-mediated trimeric coronin 1 molecules. Mol. Biol. Cell. 16:2786–2798.
Haraldsson M.K., Louis-Dit-Sully C.A., Lawson B.R., Sternik G., Santiago-Raber M.L., Gascoigne N.R., Theofilopoulos A.N., and Kono D.H. 2008. The lupus-related Lmb3 locus contains a disease-suppressing Coronin-1A gene mutation. Immunity. 28:40–51.
Kammerer R.A., Kostrewa D., Progias P., Honnappa S., Avila D., Lustig A., Winkler F.K., Pieters J., and Steinmetz M.O. 2005. A conserved trimerization motif controls the topology of short coiled coils. Proc. Natl. Acad. Sci. USA. 102:13891–13896.
Mace E.M. and Orange J.S. 2014. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proc. Natl. Acad. Sci. USA. 111:6708–6713.
Moshous D., Martin E., Carpentier W., Lim A., Callebaut I., Canioni D., Hauck F., Majewski J., Schwartzentruber J., Nitschke P., Sirvent N., Frange P., Picard C., Blanche S., Revy P., Fischer A., Latour S., Jabado N., and de Villartay J.P. 2013. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J. Allergy Clin. Immunol. 131:1594–1603.e9.
Mueller P., Massner J., Jayachandran R., Combaluzier B., Albrecht I., Gatfield J., Blum C., Ceredig R., Rodewald H.R., Rolink A.G., and Pieters J. 2008. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat. Immunol. 9:424–431.
Mueller P., Liu X., and Pieters J. 2011. Migration and homeostasis of naive T cells depends on coronin 1-mediated prosurvival signals and not on coronin 1-dependent filamentous actin modulation. J. Immunol. 186:4039–4050.
Mugnier B., Nal B., Verthuy C., Boyer C., Lam D., Chasson L., Nieoullon V., Chazal G., Guo X.J., He H.T., Rueff-Juy D., Alcover A., and Ferrier P. 2008. Coronin-1A links cytoskeleton dynamics to TCRαβ-induced cell signaling. PLoS ONE. 3:e3467.
Oku T., Itoh S., Okano M., Suzuki A., Suzuki K., Nakajin S., Tsuji T., Nauseef W.M., and Toyoshima S. 2003. Two regions responsible for the actin binding of p57, a mammalian coronin family actin-binding protein. Biol. Pharm. Bull. 26:409–416.
Punwani D., Pelz B., Yu J., Arva N.C., Schafernak K., Kondratowicz K., Makhija M., and Puck J.M. 2015. Coronin-1A: Immune deficiency in humans and mice. J. Clin. Immunol. 35:100–107.
Rybakin V. and Clemen C.S. 2005. Coronin proteins as multifunctional regulators of the cytoskeleton and membrane trafficking. BioEssays. 27:625–632.
Shiow L.R., Roadcap D.W., Paris K., Watson S.R., Grigorova I.L., Lebet T., An J., Xu Y., Jenne C.N., Foger N., Sorensen R.U., Goodnow C.C., Bear J.E., Puck J.M., and Cyster J.G. 2008. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat. Immunol. 9:1307–1315.
Shiow L.R., Paris K., Akana M.C., Cyster J.G., Sorensen R.U., and Puck J.M. 2009. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a coronin-1A mutation and a chromosome 16p11.2 deletion. Clin. Immunol. 131:24–30.
Stavropoulos, D.J., Merico, D., Jobling, R., Bowdin, S., Monfared, N., Thiruvahindrapuram, B., et al. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genom Med 2016; 1.
Stray-Pedersen A., Jouanguy E., Crequer A., Bertuch A.A., Brown B.S., Jhangiani S.N., Muzny D.M., Gambin T., Sorte H., Sasa G., Metry D., Campbell J., Sockrider M.M., Dishop M.K., Scollard D.M., Gibbs R.A., Mace E.M., Orange J.S., Lupski J.R., Casanova J.L., and Noroski L.M. 2014. Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. J. Clin. Immunol. 34:871–890.
Suzuki K., Nishihata J., Arai Y., Honma N., Yamamoto K., Irimura T., and Toyoshima S. 1995. Molecular cloning of a novel actin-binding protein, p57, with a WD repeat and a leucine zipper motif. FEBS Lett. 364:283–288.
Tchang V.S., Mekker A., Siegmund K., Karrer U., and Pieters J. 2013. Diverging role for coronin 1 in antiviral CD4+ and CD8+ T cell responses. Mol. Immunol. 56:683–692.
Wang, K., Li, M., and Hakonarson, H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet 2010; 11:843–54
Xavier C.P., Eichinger L., Fernandez M.P., Morgan R.O., and Clemen C.S. 2008. Evolutionary and functional diversity of coronin proteins. Subcell. Biochem. 48:98–109.
Yee C.S., Massaad M.J., Bainter W., Ohsumi T.K., Foger N., Chan A.C., Akarsu N.A., Aytekin C., Ayvaz D.C., Tezcan I., Sanal O., Geha R.S., and Chou J. 2016. Recurrent viral infections associated with a homozygous CORO1A mutation that disrupts oligomerization and cytoskeletal association. J. Allergy Clin. Immunol. 137:879–888.e2.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 2 • June 2019
Pages: 52 - 60
History
Received: 4 April 2019
Accepted: 22 May 2019
Accepted manuscript online: 24 May 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AdiOvadia, Vy Hong-DiepKim, BrendaReid, HarjitDadi, AnnePham-Huy, and Chaim M.Roifman. 2019. Successful hematopoietic stem cell transplantation in a patient with a novel mutation in coronin 1A. LymphoSign Journal.
6(2): 52-60. https://doi.org/10.14785/lymphosign-2019-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
