A novel homozygous mutation in CIITA resulting in MHC Class II deficiency in an adult patient
Abstract
Introduction: Major histocompatibility (MHC) class II deficiency is a rare autosomal recessive primary immunodeficiency with fewer than 200 patients reported worldwide. Patients usually present within their first year of life with severe and recurrent infections, failure to thrive, and chronic diarrhea. The disorder is caused by absent or reduced MHC class II expression on cell surfaces, leading to defective cellular and humoral immune responses. The disease is associated with a poor prognosis, with most patients dying in early childhood due to infectious complications.
Aim: To report the clinical, immunological, and genetic features of an adult patient with MHC class II deficiency who did not undergo hematopoietic stem cell transplant (HSCT). We also explore proposed theories as to why some patients with MHC class II deficiency survive to adulthood, beyond the typical life expectancy.
Results: We present a 23-year-old gentleman who was diagnosed with MHC class II deficiency at the age of 6 months based on a near complete absence of Human Leukocyte Antigen - DR isotype on peripheral blood mononuclear cells and CD4+ lymphopenia. He is one of a few patients with the condition reported in the literature to have survived to adulthood despite not having undergone HSCT. Next generation sequencing revealed a novel homozygous mutation in the CIITA gene, 1 of 4 genes involved in the regulation of MHC class II transcription.
Discussion: MHC class II deficiency is considered a single entity phenotypic condition where the main problem lies in reduced or absent MHC class II expression and results in downstream immunologic effects, including CD4+ lymphopenia and impaired antigen specific responses. However, phenotypic differences between patients are emerging as more cases are described in the literature. Our patient, now 23 years old, has survived significantly beyond life expectancy despite not having HSCT.
Statement of novelty: We describe a case of an adult patient diagnosed with MHC class II deficiency due to a novel homozygous intronic splice site variant in the CIITA gene.
Background
Major histocompatibility (MHC) class II deficiency is a rare autosomal recessive primary immunodeficiency with fewer than 200 patients reported worldwide (Ben-Mustapha et al. 2013). Most of these reported cases have been observed in North African populations, although some cases have been observed in other populations, especially in those of high consanguinity (Villard et al. 2001; Ouederni et al. 2011; Aluri et al. 2018). Patients usually present within the first year of life with severe and recurrent infections, failure to thrive, and chronic diarrhea.
The disorder is caused by absent or reduced MHC class II expression on cell surfaces. The lack of MHC class II leads to defective antigen presentation, thereby leading to impaired CD4+ T-cell development and activation, and impaired T-helper cell-dependent antibody production by B cells. Ultimately, both cellular and humoral immune responses to foreign antigens are affected.
The defect is not in the MHC class II genes themselves but rather in any one of the 4 regulatory genes involved in MHC class II gene transcription: CIITA (class II transactivator), RFXANK (regulatory factor × associated ankyrin containing protein), RFX5 (regulatory factor × 5) and RFXAP (regulatory factor × associated protein) (Villard et al. 2001; Hanna and Etzioni 2014). Mutations in these regulator genes are respectively classified into what are known as complementation groups A, B, C, D (Villard et al. 2001; Hanna and Etzioni 2014).
Due to the lack of antigen-specific immune responses, patients with MHC class II deficiency are susceptible to severe and recurrent fungal, bacterial, viral and protozoal infections, primarily affecting the respiratory and gastrointestinal tracts (Villard et al. 2001; Hanna and Etzioni 2014). Because the MHC class II molecule plays a critical role in negative thymic selection of CD4+ T-cells, MHC class II deficiency is also associated with a higher risk of autoimmunity manifesting as autoimmune cytopenias and sclerosing cholangitis (Hanna and Etzioni 2014). The disease is associated with a poor prognosis, with most patients dying in early childhood due to infectious complications.
Hematopoietic stem cell transplant (HSCT) is the only therapeutic option for cure, but its success rate is lower than in other primary immunodeficiencies even when an HLA-matched donor is available (Klein et al. 1995; Renella et al. 2006).
There are few reports of patients surviving until adulthood without HSCT (Quan et al. 1999; Wiszniewski et al. 2001; Prod’homme et al. 2003; Ouederni et al. 2011). We report on the experience of a now 23-year-old male patient of Palestinian descent with MHC class II deficiency who was treated in Canada and did not undergo hematopoietic stem cell transplant. Genetic analysis revealed a novel homozygous mutation in the CIITA gene. We also review the literature for other adults with the condition and theories as to why some patients may have a longer survival than others.
Case presentation
We present patient DH, now a 23-year-old gentleman who was diagnosed with MHC class II deficiency at the age of 6 months, when he was hospitalized with bilateral interstitial pneumonia presumed to be Pneumocystis jiroveci pneumonia. The family history was notable for consanguineous parents of Palestinian descent. The diagnosis was made based on a near complete absence of Human Leukocyte Antigen - DR isotype (HLA-DR) on peripheral blood mononuclear cells, and CD4+ lymphopenia. His family chose not to perform hematopoietic stem cell transplant because his biological sister and cousin, both affected with the same disorder, had died after transplantation. To prevent infections, he was started on monthly infusions of IVIG and antibiotics for prophylaxis against P. jiroveci pneumonia.
Concerns since infancy have been failure to thrive, malabsorption, and chronic diarrhea of unclear cause. The patient required a gastric tube for supplementary nutrition at around age 9 until age 20 when the gastric tube was removed at his request. However, dysphagia from increasingly difficult-to-treat oral and esophageal candidiasis made it challenging for him to maintain adequate oral intake. The gastric tube was reinserted after a drop in weight from a highest of 40 kg down to 34 kg.
In terms of infections, in childhood, DH had recurrent ear infections and recurrent gastrointestinal infections with Clostridium difficile, Giardia intestinalis, and rotavirus. He was frequently hospitalized for febrile neutropenia and pneumonias caused by Coronavirus, Rhinovirus and Pseudomonas pneumoniae. By age 8, he developed bronchiectasis. As mentioned, he continues to have chronic and persistent oral and esophageal candidiasis with Candida glabrata and Candida albicans, which has been difficult to treat due to drug resistance. Up to age 20, his infections responded well to oral and parenteral antibiotics and required only short admissions to hospital.
Over the past year, his health has deteriorated with nearly monthly admissions to hospital for recurrent pneumonias, episodes of febrile neutropenia, and autoimmune hemolytic anemia requiring blood transfusions. He has had recent lung colonization with P. aeruginosa. Recently, he has had a trend of increasingly elevated liver enzymes in a cholestatic pattern and hepatomegaly, causing concerns of progressive hepatic dysfunction. Our work-up has not yet been able to elucidate the cause of his liver dysfunction, though Cryptosporidium infection and sclerosing cholangitis is often implicated as a cause of progressive liver dysfunction in other reported cases (Hanna and Etzioni 2014).
Immunologic findings for our patient are summarized in Table 1, which mainly shows CD4+ lymphopenia, and a profound deficit of HLA-DR expression on lymphocytes.
Table 1:
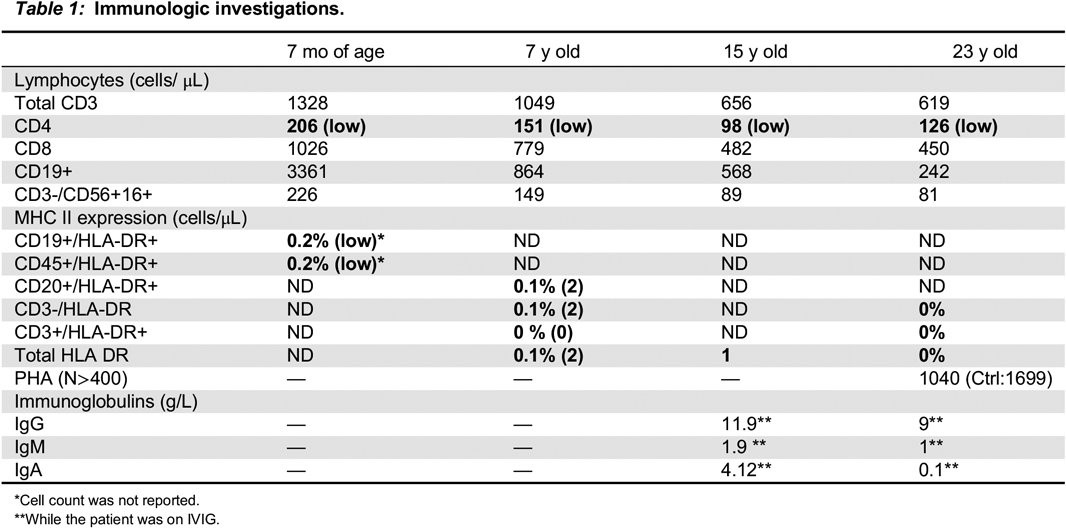
*
Cell count was not reported.
**
While the patient was on IVIG.
He continues to be treated supportively with intravenous immunoglobulin (IVIG) therapy every 3 weeks, trimethoprim-sulfamethoxazole for prophylaxis against P. jiroveci pneumonia, and clotrimazole for recurrent oral and esophageal candidiasis. Apart from his long survival with this disorder without HSCT, his clinical features and immunologic findings are consistent with what is described in the literature.
Genetic evaluation
Sequence analysis (Primary Immunodeficiency Panel, Blueprint Genetics) revealed a novel homozygous intronic splice site variant in CIITA, resulting in c.3317+2dup. This mutation is predicted to weaken the natural splice donor site leading to aberrant splicing as well as skipping of exon 18.
Discussion
MHC class II deficiency is considered a single entity phenotypic condition in that patients have reduced or absent MHC class II expression, and patients generally share some of the “classic”, expected downstream immunologic and clinical consequences of this defect, such as CD4+ lymphopenia, and impaired antigen specific responses. However, phenotypic differences between patients are emerging as more cases are described in the literature. For example, the condition was originally described as a disease that can be lethal in childhood, but there are reported cases of adults in their twenties and thirties (see Table 2), including our patient DH (Quan et al. 1999; Wiszniewski et al. 2001; Towey and Kelly 2002; Prod’homme et al. 2003; Ouederni et al. 2011). There are also reports of patients who present with mild symptoms. These observations have an important implication in that that there may be factors at play in these patients that can prolong survival even without HSCT.
Table 2:
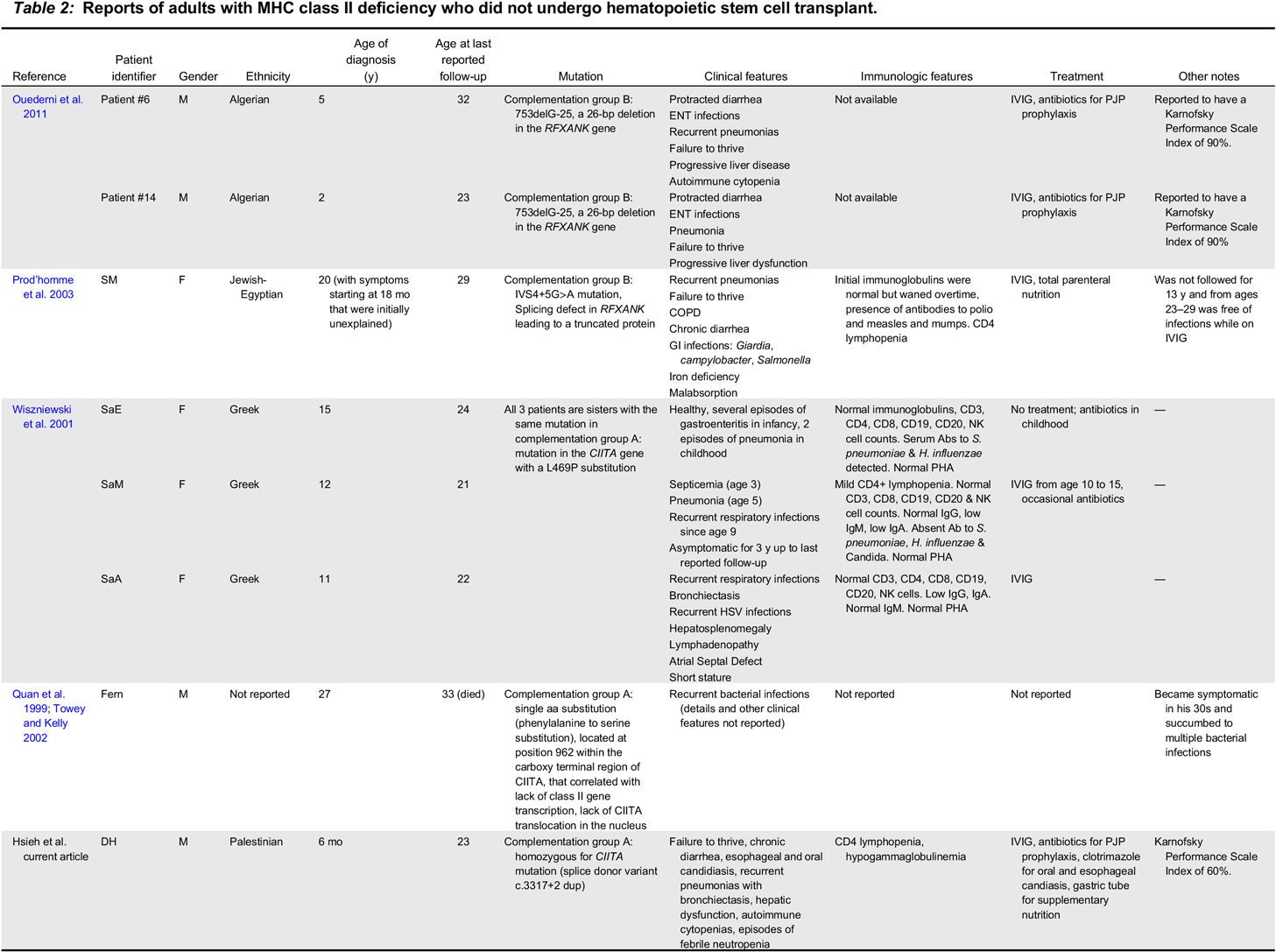
Several theories have been proposed to explain phenotypic differences such as longer survival and milder disease course. One theory is that there is some degree of residual activity of the mutant regulator protein. For example, in a report by Wiszniewski et al. (2001), 3 sisters, the Sa sisters, with mild/asymptomatic disease with a homozygous missense mutation in CIITA were found to have faint expression of HLA-DP, -DR, and -DQ on B cells and monocytes, leading one to speculate that there may be some residual functional activity in the mutated CIITA gene (Wiszniewski et al. 2001). When the mutant gene was transfected in a CIITA-deficient cell line, HLA-DR expression was restored in 30% of cells (Wiszniewski et al. 2001). Interestingly, they also found that the mutated protein is able to translocate into the nucleus whereas other known CIITA mutated proteins cannot (Wiszniewski et al. 2001). As was reported on the Sa sisters, our patient, DH, did not have complete absence of MHC class II molecules. Instead, he had near absent, but detectable expression of HLA-DR on 0.1% of T and B cells meaning there could be residual activity of the defective CIITA regulator protein. We will need to follow-up with functional analyses of DH’s mutation to explore how this defective regulatory protein behaves. But, if indeed the mutation that DH has leads to a defective regulatory protein with partial activity, it could be one explanation as to why his sister, who presumably had the same mutation, may have died from complications with HSCT. Engraftment failure due to residual adaptive immunity has been cited as one of the major reasons why HSCT in this population has had only limited success in the past (Klein et al. 1995; Renella et al. 2006; Gennery et al. 2010).
There may be other factor(s) apart from residual activity that explains the differences in immunologic phenotype that we have yet to explain. Ouederni et al. (2011) reported on 35 patients of North African descent with the same genetic mutation, a homozygous I5E6-25_15E6 +1 deletion in RFXANK. It is a mutation seen in approximately 70% of patients with MHC class II deficiency and was traced to an ancestor belonging to the Berber civilization who lived 2250 years ago (Ouederni et al. 2011). Despite having the same genetic mutation, there was wide variability in their phenotypes. For example, while many patients had died in childhood due to infectious complications or from complications after HSCT, 4 of 12 patients who did not undergo HSCT had reached puberty, and of these 4 patients, only 1 patient displayed residual MHC class II expression on B cells (Ouederni et al. 2011). For the other 3 patients, is unclear what factors would have influenced their survival.
Another theory that has not been well explored is the possible presence of immune compensatory mechanisms that make up for the loss of MHC class II expression, for example upregulation of MHC I or innate immunity (Prod’homme et al. 2003). Furthermore, there could be external factors such access to medical care, hygienic practices, social support, and other environmental factors (Prod’homme et al. 2003; Ouederni et al. 2011).
In summary, we present a case of a 23-year-old gentleman with MHC class II deficiency with a novel c.3317+2dup homozygous CIITA mutation. Based on a review of the literature, there are other cases of MHC class II deficient patients who survive into adulthood, significantly beyond the expected life expectancy even without HSCT. We can only conjecture why there is this variability in immunologic phenotype given the small number of patients reported in the literature, but there are likely to be undefined factors that have a major impact on the severity and course of the disease.
REFERENCES
Aluri J., Gupta M., Dalvi A., Mhatre S., Kulkarni M., Hule G., Desai M., Shah N., Taur P., Vedam R., and Madkaikar M. 2018. Clinical, immunological, and molecular findings in five patients with major histocompatibility complex class II deficiency from India. Front. Immunol. 9:188.
Ben-Mustapha I., Ben-Farhat K., Guirat-Dhouib N., Dhemaied E., Larguèche B., Ben-Ali M., Chemli J., Bouguila J., Ben-Mansour L., Mellouli F., Khemiri M., Béjaoui M., and Barbouche M.R. 2013. Clinical, immunological and genetic findings of a large Tunisian series of major histocompatibility complex class II deficiency patients. J. Clin. Immunol. 33(4):865–870.
Gennery A.R., Slatter M.A., Grandin L., Taupin P., Cant A.J., Veys P., Amrolia P.J., Gaspar H.B., Davies E.G., Friedrich W., Hoenig M., Notarangelo L.D., Mazzolari E., Porta F., Bredius R.G., Lankester A.C., Wulffraat N.M., Seger R., Güngör T., Fasth A., Sedlacek P., Neven B., Blanche S., Fischer A., Cavazzana-Calvo M., Landais P., and Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation European Society for Immunodeficiency. 2010. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: Entering a new century, do we do better? J. Allergy Clin. Immunol. 126(3):602–610.e1–11.
Hanna S. and Etzioni A. 2014. MHC class I and II deficiencies. J. Allergy Clin. Immunol. 134(2):269–275.
Klein C., Cavazzana-Calvo M., Le Deist F., Jabado N., Benkerrou M., Blanche S., Lisowska-Grospierre B., Griscelli C., and Fischer A. 1995. Bone marrow transplantation in major histocompatibility complex class II deficiency: A single-center study of 19 patients. Blood. 85(2):580–587.
Ouederni M., Vincent Q.B., Frange P., Touzot F., Scerra S., Bejaoui M., Bousfiha A., Levy Y., Lisowska-Grospierre B., Canioni D., Bruneau J., Debré M., Blanche S., Abel L., Casanova J.L., Fischer A., and Picard C. 2011. Major histocompatibility complex class II expression deficiency caused by a RFXANK founder mutation: A survey of 35 patients. Blood. 118(19):5108–5118.
Prod’homme T., Dekel B., Barbieri G., Lisowska-Grospierre B., Katz R., Charron D., Alcaide-Loridan C., and Pollack S. 2003. Splicing defect in RFXANK results in a moderate combined immunodeficiency and long-duration clinical course. Immunogenetics. 55(8):530–539.
Quan V., Towey M., Sacks S., and Kelly A.P. 1999. Absence of MHC class II gene expression in a patient with a single amino acid substitution in the class II transactivator protein CIITA. Immunogenetics. 49(11–12):957–963.
Renella R., Picard C., Neven B., Ouachée-Chardin M., Casanova J.L., Le Deist F., Cavazzana-Calvo M., Blanche S., and Fischer A. 2006. Human leucocyte antigen-identical haematopoietic stem cell transplantation in major histocompatiblity complex class II immunodeficiency: Reduced survival correlates with an increased incidence of acute graft-versus-host disease and pre-existing viral infections. Br. J. Haematol. 134(5):510–516.
Towey M. and Kelly A.P. 2002. Nuclear localisation of CIITA is controlled by a carboxy terminal leucine-rich repeat region. Mol. Immunol. 38(8):627–634.
Villard J., Masternak K., Lisowska-Grospierre B., Fischer A., and Reith W. 2001. MHC class II deficiency: A disease of gene regulation. Medicine (Baltimore). 80(6):405–418.
Wiszniewski W., Fondaneche M.C., Le Deist F., Kanariou M., Selz F., Brousse N., Steimle V., Barbieri G., Alcaide-Loridan C., Charron D., Fischer A., and Lisowska-Grospierre B. 2001. Mutation in the class II trans-activator leading to a mild immunodeficiency. J. Immunol. 167(3):1787–1794.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 4 • December 2018
Pages: 135 - 140
History
Received: 13 October 2018
Accepted: 18 November 2018
Version of record online: 21 December 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
JaneHsieh, AmarillaMandola, and Stephen D.Betschel. 2018. A novel homozygous mutation in CIITA resulting in MHC Class II deficiency in an adult patient. LymphoSign Journal.
5(4): 135-140. https://doi.org/10.14785/lymphosign-2018-0015
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
