Interleukin-2 receptor common gamma chain (IL2RG) defects present a diagnostic challenge
Abstract
Background: The protein encoded by interleukin-2 receptor common gamma chain (IL2RG) is an important signaling component of many interleukin receptors, including those of interleukin-2, -4, -7, and -21, known as the common gamma chain. Mutations in the gene encoding the common gamma chain of the interleukin-2 receptor cause X-linked severe combined immunodeficiency (SCID). In this report, we present an unknown genetic defect of a patient diagnosed with SCID whose genetic analysis was performed 2 decades later.
Methods: Whole genome sequencing and Sanger confirmation were used to identify a novel frameshift mutation in IL2RG. Massively parallel sequencing of genes associated with SCID were performed on the patient’s mother and sister.
Results: Next generation sequencing techniques identified a heterozygous frame-shift deletion in the gene encoding the common gamma chain of IL2RG in our patient. The patient’s mother had a low level mosaicism for the same deletion. The sister had no detectable deletion.
Conclusion: We have identified a novel mutation in IL2RG resulting in an X-linked SCID phenotype. The genetic analysis of the patient’s mother revealed a mosaicism which was not passed on to his sister. The importance of genetic analysis in family members and SCID patients with an unknown genetic defect should be emphasized for family planning and subsequent genetic counseling.
Statement of novelty: Genetic testing is an extremely important component in evaluating severe combined immunodeficiency as it impacts treatment course and prognosis, and allows for genetic analysis and counselling of family members.
Background
Severe combined immunodeficiency (SCID) represents a rare group of primary immunodeficiency disorders (PIDs) characterized by a reduced number of T lymphocytes in association with a functional or quantitative defect in B lymphocytes, natural killer cells, or both. Patients with SCID may have known or yet unidentified genetic alterations explaining their phenotype. Mutations in the IL2RG gene, which encodes the common gamma chain of the interleukin-2 receptor, cause X-linked SCID (X-SCID) as well as X-linked combined immunodeficiency and remain the most common cause of SCID. This protein is an important signaling component of many interleukin receptors, including those of interleukin-2, -4, -7, and -21, and has therefore been commonly known as the common gamma chain. More than 300 mutations in the IL2RG gene have been identified in X-SCID. These mutations are located on the long arm of the X chromosome at position 13.1 (Shearer et al. 2014; Pérez-Simón 2015). The following mutational hot spots in IL2RG have been reported, at codons p.Arg224, p.Arg226, p.Arg285, and p.Arg289 (Puck et al. 1997).
Transplacental maternal engraftment (TME) is defined as the presence of maternal T cells in peripheral blood before bone marrow transplantation. The human placenta allows for bidirectional passage of nucleated cells between mother and fetus, and in healthy infants the immune system eradicates maternal cells. Patients with SCID lack the functional immunity required to reject circulating maternal T cells, resulting in persistent TME in up to 40% of these patients (Fischer et al. 1997; Liu et al. 2016). Although TME can be asymptomatic, some infants with SCID and TME can have clinical symptoms of graft-versus-host disease (GvHD) at diagnosis. The presentation of maternal engraftment can range from a fine maculopapular erythema to generalized erythroderma and alopecia. Liver, gastrointestinal, and hematologic involvement may be observed (Wahlstrom et al. 2017). A newly diagnosed patient with SCID presenting with detectable T cells is evaluated for chimerism by HLA typing of T cells and non-T cells (Müller et al. 2001). In patients with engraftment, maternal T cells are characterized by phenotype and function in response to mitogen stimulation (Müller et al. 2001). The presence of TME may persist post-transplantation, in which the patient should be assessed for signs of acute GvHD with repeat phenotypic and functional T cell analysis (Wahlstrom et al. 2017). TME has also been an impediment to proper infant immune evaluation as well as genetic analysis. Genetic testing is extremely important in SCID as early diagnosis allows for life-saving interventions such as bone marrow transplantation, which results in a higher survival rate when administered during the first 3 months of life (Kwan et al. 2014; Wahlstrom et al. 2015). In addition, proper molecular diagnosis aids in the important task of family genetic counseling.
Here, we present a patient with TME that posed a challenge to both genetic diagnosis and genetic counselling, and whose evaluation resulted in the identification of a novel mutation.
Methods
Patient
Following informed consent, patient information was collected from medical records in accordance with REB Protocol No. 1000005598.
Sanger sequencing
Genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers. Sequencing was performed using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Next generation sequencing
Massively parallel sequencing was performed on a panel of 20 SCID genes (ADA, AK2, CARD11, CD247, CD3D, CD3E, DCLRE1C, IL2RG, IL7R, JAK3, LIG4, NHEJ1, PNP, PRKDC, PTPRC, RAC2, RAG1, RAG2, RMRP, ZAP70), following standard procedures for DNA sample preparation. Libraries were constructed using the Kapa Hyper Prep kit, and targeted capture of coding exons as well as splice junctions performed with Nimblegen’s SeqCap EZ Choice. Sequencing was done with 150 bp paired-end reads on an Illumina MiSeq.
Case presentation
Our patient, now 25 years old, was diagnosed with SCID at 5 months of age. His clinical history was significant for maternal engraftment, failure to thrive and recurrent infections including pneumonitis and pneumocystis jiroveci. Pertinent laboratory workup revealed a low mitogenic response and the lymphocyte subsets were irrelevant due to maternal engraftment. His parents were of English decent. He was one of the first patients world-wide to receive a matched unrelated bone marrow transplantation at 1 year of age. His conditioning consisted of Busulfan and Cyclophosphamide. He had an uneventful transplant course with the exception of mild cutaneous GvHD. His engraftment was full and rapid with no complications. He continues to do extremely well 2 decades later, with no episodes of infections, autoimmunity, or atopy. His engraftment remains solid and immune reconstitution is complete.
Genetic analysis
Sanger sequencing of patient’s peripheral blood mononuclear cells performed in the early 90s detected no abnormalities in the IL2RG, ADA, and RAG1/2 genes, likely because of TME. Several years later, a more extensive panel of SCID-causing genes was sequenced using patient-derived EBV transformed cell lines, a DNA source not impacted by TME. This time, a novel single base deletion in IL2RG, c.245delC causing a frameshift mutation (p.Pro82fs) was identified, as demonstrated in Figure 1. However, because transformed lines are notorious for EBV-induced genetic aberrations this finding could not have been used as a definitive diagnosis. Sanger sequencing of maternal cells was normal for IL2RG, suggesting this might have been either a de-novo mutation or false-positive result. The next option for diagnosis was to obtain fibroblasts via a skin biopsy, or epithelial cells from a buccal smear.
Figure 1:
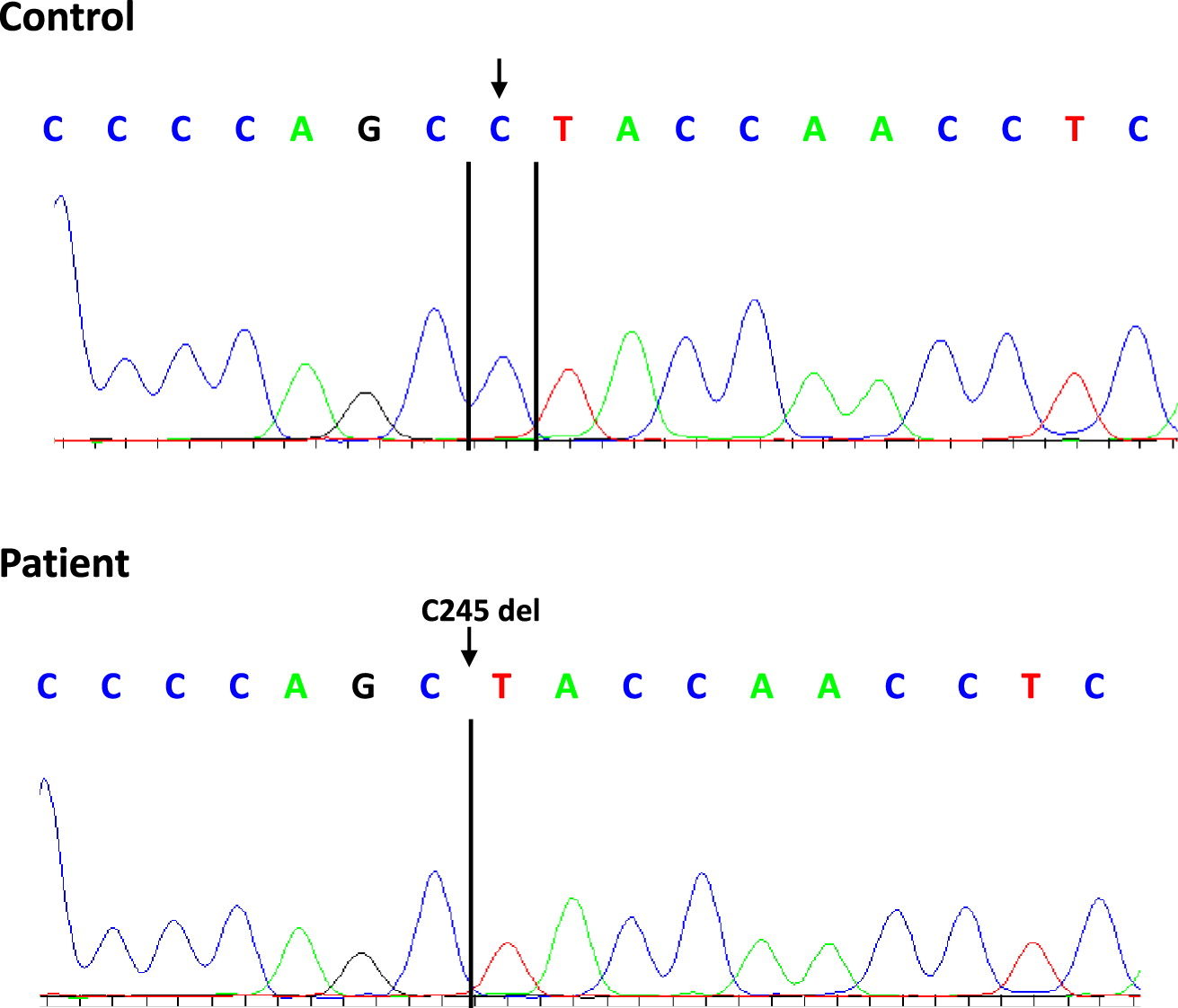
Next generation sequencing (Stavropoulos et al. 2016) performed on a buccal smear showed that approximately 81% of the sequence contained the deletion in IL2RG and 19% of the sequence was wild-type. This result could be consistent with the fact that buccal-derived cells can be contaminated with engrafted donor cells, likely stemming from lymphocytes.
Sequencing performed in our patient’s sister did not detect the single base deletion in IL2RG. A similar analysis on the patient’s mother yielded 1 read out of 164 that showed the same deletion. This is an extremely low level of mosaicism and could be easily missed by performing traditional sequencing.
Discussion
Patients with SCID may have unknown genetic mutations explaining their phenotype. Gene defects account for approximately 85% of SCID cases. Prior to newborn screening and in the absence of family history of SCID, most patients came to medical attention with failure to thrive, recurrent and opportunistic infections, such as our case. Over the years, genetic analysis has become an important component of an evaluation of a patient with SCID as it impacts prenatal diagnosis, treatment course, and prognosis. Further, the importance of genetic analysis in family members of such patients needs to be emphasized for family planning and subsequent genetic counselling. Our patient underwent next generation sequencing (SCID panel) 2 decades following his SCID diagnosis, confirming a suspected mutation in IL2RG. Subsequently, his mother and sister underwent molecular genetic testing which revealed mosaicism (<1%) in the mother and no deletion in the sister. This extremely low level of mosaicism was the result of a post-zygotic mutation in the mother, hence only some of her cells were affected. As the sister had no deletion detected, her risk of being mosaic was extremely low and she is therefore unlikely to pass on the mutation to her children.
Our patient poses no risk to passing on his mutation to his male children given his X-linked condition, but his risk of passing on the mutation to his female children is 100%. Thus, none of the patient’s offspring will develop SCID, but female descendants will be carriers of the pathogenic variant.
This case report describes a novel mutation in IL2RG, emphasizing the complexity of genetic analysis in SCID patients and their family members, and the importance of pursuing a molecular diagnosis. Next generation sequencing appears superior to traditional methods in providing answers for family planning and subsequent genetic counselling.
REFERENCES
Fischer A., Cavazzana-Calvo M., De Saint Basile G., DeVillartay J.P., Di Santo J.P., Hivroz C., Rieux-Laucat F., and Le Deist F. 1997. Naturally occurring primary deficiencies of the immune system. Annu. Rev. Immunol. 15:93–124.
Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., Baker M., Ballow M., Bartoshesky L.E., Bonilla F.A., Brokopp C., Brooks E., Caggana M., Celestin J., Church J.A., Comeau A.M., Connelly J.A., Cowan M.J., Cunningham-Rundles C., Dasu T., Dave N., De La Morena M.T., Duffner U., Fong C.T., Forbes L., Freedenberg D., Gelfand E.W., Hale J.E., Hanson I.C., Hay B.N., Hu D., Infante A., Johnson D., Kapoor N., Kay D.M., Kohn D.B., Lee R., Lehman H., Lin Z., Lorey F., Abdel-Mageed A., Manning A., McGhee S., Moore T.B., Naides S.J., Notarangelo L.D., Orange J.S., Pai S.Y., Porteus M., Rodriguez R., Romberg N., Routes J., Ruehle M., Rubenstein A., Saavedra-Matiz C.A., Scott G., Scott P.M., Secord E., Seroogy C., Shearer W.T., Siegel S., Silvers S.K., Stiehm E.R., Sugerman R.W., Sullivan J.L., Tanksley S., Tierce M.L. IV, Verbsky J., Vogel B., Walker R., Walkovich K., Walter J.E., Wasserman R.L., Watson M.S., Weinberg G.A., Weiner L.B., Wood H., Yates A.B., Puck J.M., and Bonagura V.R. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 312:729–738.
Liu C., Duffy B., Bednarski J.J., Calhoun C., Lay L., Rundblad B., Payton J.E., and Mohanakumar T. 2016. Maternal T-cell engraftment interferes with human leukocyte antigen typing in severe combined immunodeficiency. Am. J. Clin. Pathol. 145:251–257.
Müller S.M., Ege M., Pottharst A., Schulz A.S., Schwarz K., and Friedrich W. 2001. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: A study of 121 patients. Blood. 98(6):1847–1851.
Pérez-Simón J.A. 2015. Anti-common γ-chain antibody: One for all in GVHD. Blood. 125(3):424–426.
Puck J.M., Pepper A.E., Henthorn P.S., Candotti F., Isakov J., Whitwam T., Conley M.E., Fischer R.E., Rosenblatt H.M., Small T.N., and Buckley R.H. 1997. Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood. 89:1968–1977.
Shearer W.T., Dunn E., Notarangelo L.D., Dvorak C.C., Puck J.M., Logan B.R., Griffith L.M., Kohn D.B., O’Reilly R.J., Fleisher T.A., Pai S.-Y., Martinez C.A., Buckley R.H., and Cowan M.J. 2014. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: The Primary Immune Deficiency Treatment Consortium experience. J. Allergy Clin. Immunol. 133:1092–1098.
Stavropoulos D.J., Merico D., Jobling R., Bowdin S., Monfared N., Thiruvahindrapuram B., Nalpathamkalam T., Pellecchia G., Yuen R.K.C., Szego M.J., Hayeems R.Z., Shaul R.Z., Brudno M., Girdea M., Frey B., Alipanahi B., Ahmed S., Babul-Hirji R., Porras R.B., Carter M.T., Chad L., Chaudhry A., Chitayat D., Doust S.J., Cytrynbaum C., Dupuis L., Ejaz R., Fishman L., Guerin A., Hashemi B., Helal M., Hewson S., Inbar-Feigenberg M., Kannu P., Karp N., Kim R., Kronick J., Liston E., MacDonald H., Mercimek-Mahmutoglu S., Mendoza-Londono R., Nasr E., Nimmo G., Parkinson N., Quercia N., Raiman J., Roifman M., Schulze A., Shugar A., Shuman C., Sinajon P., Siriwardena K., Weksberg R., Yoon G., Carew C., Erickson R., Leach R.A., Klein R., Ray P.N., Meyn M.S., Scherer S.W., Cohn R.D., and Marshall C.R. 2016. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom. Med. 1:15012.
Wahlstrom J.T., Dvorak C.C., and Cowan M.J. 2015. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr. Pediatr. Rep. 3:1–10.
Wahlstrom J.T., Patel K., Eckhert E., Kong D., Horn B., Cowan M.J., and Dvorak C.C. 2017. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J. Allergy Clin. Immunol. 139:628–633.e10.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 5 • Number 4 • December 2018
Pages: 130 - 134
History
Received: 6 July 2018
Accepted: 5 November 2018
Version of record online: 21 December 2018
Copyright
© 2018.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
CarolineWeisser, Dennis E.Bulman, KaylaFlamenbaum, and MaianRoifman. 2018. Interleukin-2 receptor common gamma chain (IL2RG) defects present a diagnostic challenge. LymphoSign Journal.
5(4): 130-134. https://doi.org/10.14785/lymphosign-2018-0009
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
