Chronic mucocutaneous candidiasis: a spectrum of genetic disorders
Abstract
Chronic mucocutaneous candidiasis (CMCC) encompasses a heterogeneous group of syndromes associated with persistent or recurrent Candida infections of the skin, nails, and mucous membranes. While chronic candidiasis can present by itself or as part of a complex, including endocrinopathy, autoimmune manifestations, bone marrow failure and neoplastic diseases, it is often regarded as a warning sign for immunodeficiency. Here, we review the processes involved in host-microbial recognition of Candida and highlight underlying genetic causes of CMCC—including those that are monogenic (such as mutations in AIRE and STAT1) as well as polymorphisms that increase susceptibility to candidal infection.
Statement of novelty: This review provides an overview of the pathophysiology of Candida fungal infection as well as genetic defects that have been identified to cause CMCC.
Introduction
Chronic mucocutaneous candidiasis (CMCC) encompasses a heterogeneous group of syndromes associated with persistent or recurrent Candida infections of the skin, nails, and mucous membranes (Kirkpatrick 1994). While chronic candidiasis can present by itself or as part of a complex, including endocrinopathy, autoimmune manifestations, bone marrow failure and neoplastic diseases, it is often regarded as a warning sign for immunodeficiency. Indeed, much current knowledge is derived from study of families and individuals with immune dysfunction.
Although the exact mechanisms underlying CMCC are still being unraveled, advancements made in whole genome sequencing techniques have been key to furthering our understanding of the pathophysiology as well genetic causes of this disorder. Here, we review the pathophysiology of Candida fungal infection as well as genetic defects that have been identified to cause CMCC.
Candida infection in healthy and immunocompromised individuals
Candida spp. are part of the commensal microflora and often found on mucosal surfaces including the mouth, intestine, vagina, and skin of healthy individuals. These microorganisms contribute to the development of the host immune system, however, can act as opportunistic pathogens when there is microbial imbalance, for example, due to immune suppression associated with antibiotic or steroid treatment. C. albicans is among the most prevalent and well-studied microorganism identified in CMCC.
Most healthy women will experience transient vaginal candidiasis at some point in their lifetime (Sobel 2007). In contrast, Candida infection in an immunocompromised individual can cause severe systemic infection, candidemia, which in turn can be fatal in a considerable number of patients (Das et al. 2011). Almost all oncologic patients with neutropenia experience oropharyngeal candidiasis (Viscoli et al. 1999), and in patients undergoing bone marrow transplantation, as many as 40% suffer systemic infections. Of these, 4% are infected with Candida and the associated mortality rate may be as high as 42% (Ortega et al. 2005).
Another risk factor for candidemia is prolonged hospitalization; such is the case for patients in intensive care (Das et al. 2011). Surgical procedures and insertion of medical devices are also associated with higher incidence of candidemia and is augmented by the ability of Candida to create biofilms on medical devices such as central vein catheters (Donlan and Costerton 2002). Neonates with central catheters experience high rates of bloodstream infections, with Candida being the third most common microbial source (Chitnis et al. 2012).
It is important to note that in most patients with candidiasis, no single genetic cause can be found. Rather, susceptibility to infection is due a combination of genetic variants (polymorphisms) and environmental factors, as mentioned above. In contrast, the underlying causes of CMCC may be monogenic (Table 1), such as single gene mutations in autoimmune regulator (AIRE), signal transducer and activator of transcription-1 (STAT1) and -3 (STAT3), or the result of polymorphisms in genes encoding Dectin-1, NACHT LRR and PYD-containing protein 3 (NLRP3), protein tyrosine phosphatase non-receptor type-22 (PTPN22), and Toll-like receptors (TLRs) which contribute to disease susceptibility (Table 2). Candidiasis in these circumstances can be a dominating, constitutive feature or an associated, inconsistent sign.
Table 1:
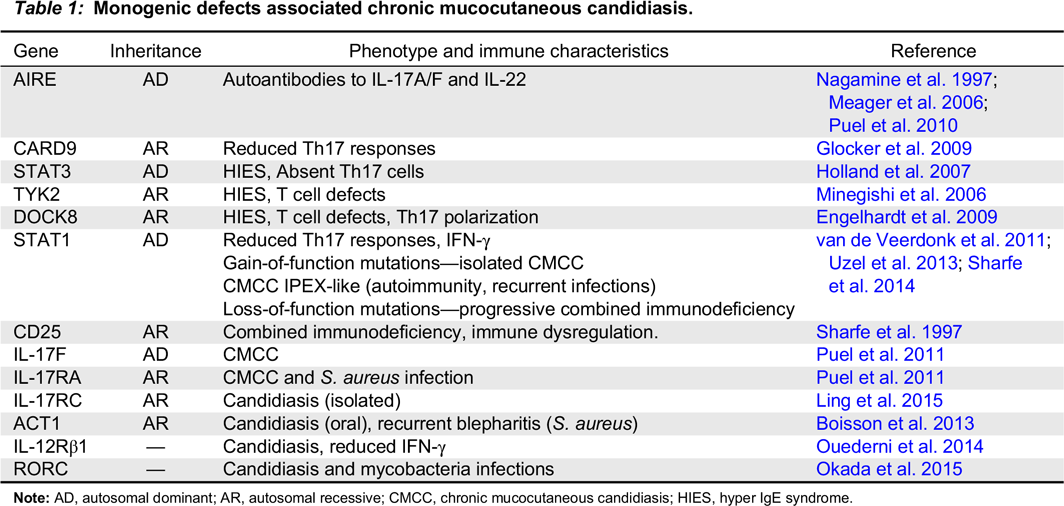
Note: AD, autosomal dominant; AR, autosomal recessive; CMCC, chronic mucocutaneous candidiasis; HIES, hyper IgE syndrome.
Table 2:
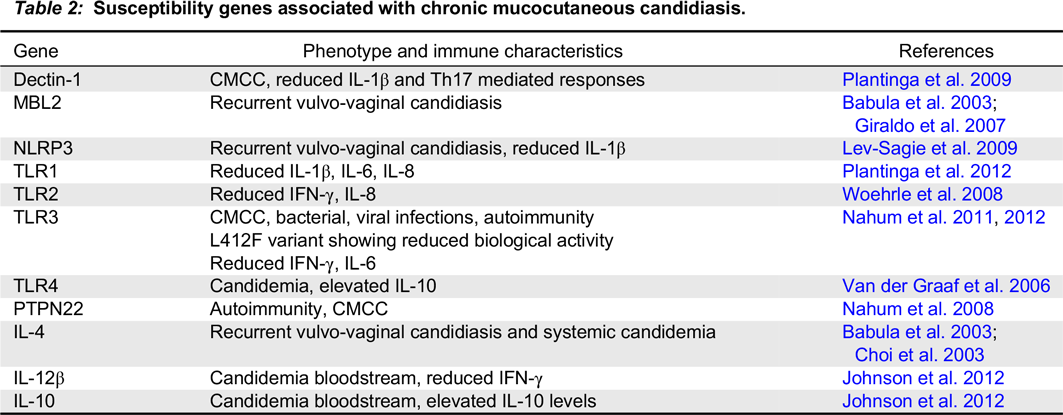
Detection of Candida by immune cells
The cell membrane of Candida consists of 2 distinct layers; an inner layer composed of polysaccharides such as chitin, 1,3-beta-glucans and 1,6-beta-glucans, and an outer layer structured mainly of proteins that are mannosylated with mannan side-chains. These components serve as ligands for receptors of the innate immune system, such as TLRs and C-type lectins (CLRs) that are found on the surface of immune cells (Smeekens et al. 2013a).
Pattern recognition receptors are requisite for host microbial recognition of Candida (Figure 1). Whereas TLR2 recognizes phospholipo-mannans (Jouault et al. 2003), TLR4 recognizes O-linked mannans (Netea et al. 2006; Ferwerda et al. 2009). CLRs such as Dectin-2 recognize mannose residues (McGreal et al. 2006) while Dectin-1 is activated by β-glucan (Brown and Gordon 2001; Rogers et al. 2005). Upon ligation, these receptors signal through different downstream adaptor molecules and kinases, resulting in secretion of pro-inflammatory cytokines. At the same time, host recognition of the pathogen ensures phagocytosis and killing of the pathogen and presentation to other immune cells.
Figure 1:
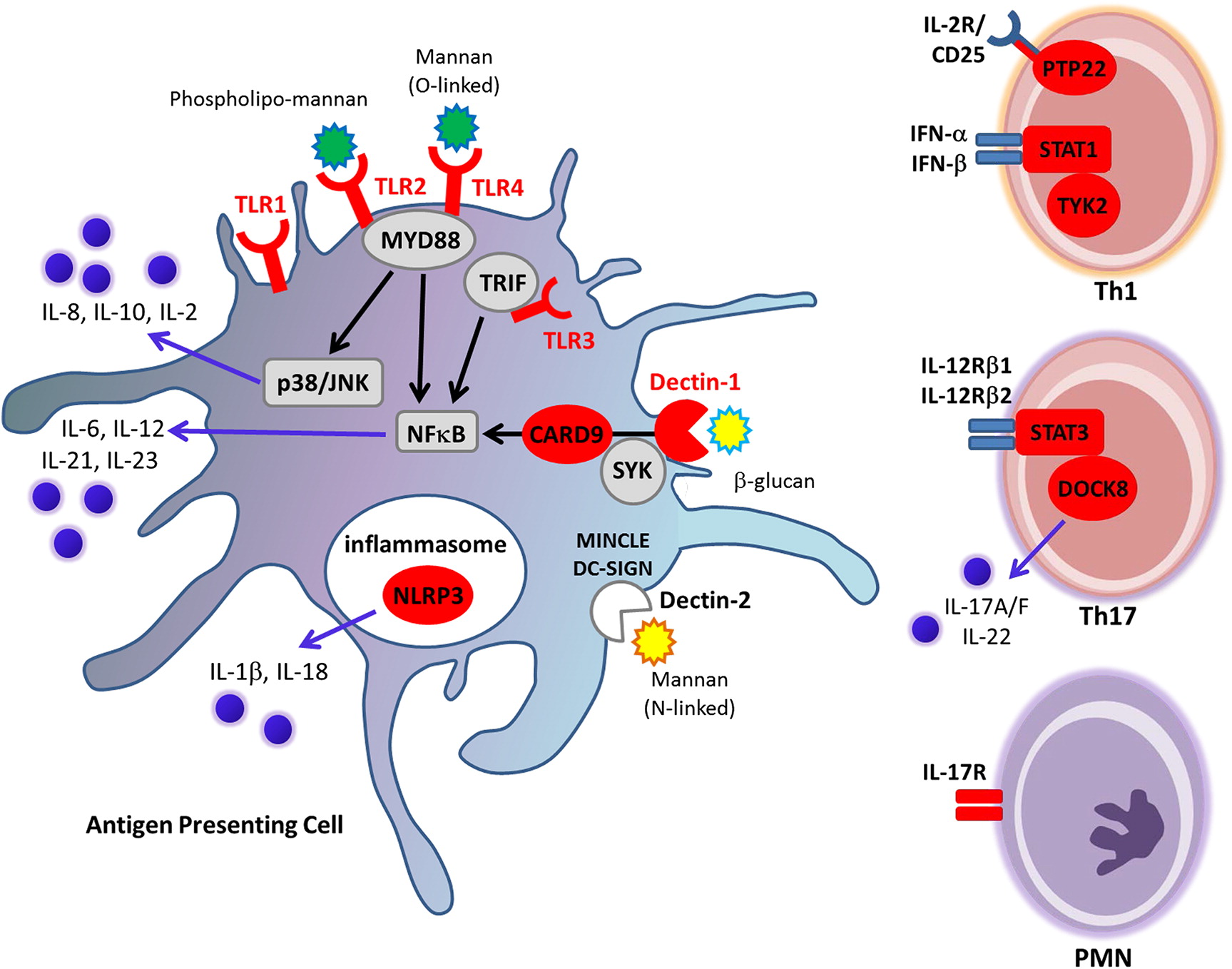
The prompt and correct recognition by these receptors also define and shape the next phase of the immune response by adaptive immune components (Akira et al. 2006). Recently, it was shown that exposure of Candida to healthy individuals induces expression of type I interferon (IFN) family cytokines, skewing the immune response towards Th1 responses. This resulted in a unique cytokine profile that was different from that induced by bacterial pathogens (Smeekens et al. 2013b).
Th1 and Th17 cell-mediated responses are central for controlling Candida infections. Their precise role was elucidated through different genetic defects and autoimmune phenomenon resulting in reduced interleukin (IL)-17F, IL-17A, and IL-22 activity alongside the occurrence of fungal infections. The differentiation of naïve T cells to Th17 cells is dependent upon a certain cytokine milieu. Upon recognition of fungal cell wall elements by pattern recognition receptors on activated T cells and antigen presenting cells, cytokines such as IL-1β, IL-6, IL-21, IL-23 and transforming growth factor (TGF)-β are secreted. These lead to activation of transcription factors STAT-1, STAT-3 and retinoic acid orphan receptor-γt (RORγt), resulting in polarization to Th17 cells which in turn exert their effects by secreting IL-17A, IL-17F, IL-22 (Ivanov et al. 2006; Yang et al. 2007). These cytokines further induce secretion of chemokines that attract neutrophils to sites of infection, and act in concert with TNF-α and IL-22 to induce anti-microbial peptide (AMP) from endothelial cells (Iwakura et al. 2011; Hernández-Santos and Gaffen 2012; McGeachy and McSorley 2012; Rutz et al. 2013).
Together, these components are necessary to control and overcome Candida infections. Aberration or immune dysregulation during any of these steps is thought to underlie CMCC.
Monogenic defects causing CMCC
Several monogenic defects are known to cause CMCC. These are described in the next paragraphs.
Autoimmune regulator (AIRE) deficiency
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) associated CMCC was the first to be described and deciphered genetically. APECED is caused by mutations in the AIRE gene, resulting in loss-of-function of the encoded AIRE transcription factor (Nagamine et al. 1997; Björses et al. 1998). It is transmitted in an autosomal recessive manner, and is found mainly in certain populations; 1:25 000 in Finns, 1:14 500 in Sardinians and 1:9000 in Iranian Jews. While more than 50 mutations have been described to date, the most common mutation is the R257X variant identified in the Finnish population. It is noteworthy, however, that the majority of patients with CMCC outside of these ethnic groups do not carry mutations in AIRE.
APECED is characterized by the classic triad of chronic candidiasis (appearing in almost all patients), adrenal failure which is present in 84% of patients, and hypoparathyroidism which presents in 88% of patients. Hypogonadism is also very common (mainly ovarian failure, and to lesser extent male hypogondasim). Such individuals may also present with other endocrynopathies; hypothyroidism and diabetes mellitus are found in as many as 1/3 of patients (Perheentupa 2006). Skin is often involved and appears dystrophic, and common skin manifestations include vitiligo and alopecia, which is found in as many as 30% of patients. Teeth enamel is also frequently affected. Physicians should stay alert for occurrence of keratoconjunctivitis which could lead to blindness. Other less common manifestations are chronic diarrhea as well as bouts of rash and fever (Perheentupa 2006). Hepatitis, ranging from asymptomatic elevated liver enzymes to severe necrotizing hepatitis, has been reported in 12 of 68 patients and pernicious anemia in 13 of 68 patients (Ahonen et al. 1990).
The AIRE transcription factor is responsible for regulating the expression of autoantigen receptors on thymocytes. During lymphocyte maturation in the thymus, the random rearrangement of antigen-specific receptor genes produces a vast and diverse repertoire of receptors, some of which mistakenly recognize “self” as “non-self”. To prevent autoimmunity, these cells undergo clonal deletion, a process that is closely regulated by AIRE in thymus medullary epithelial cells. Thus, T cells recognizing these antigens with high affinity are clonally deleted. AIRE deficiency resulting in loss-of-function thus allows T cell expressing autoantigen receptors to expand to the periphery and induce autoimmunity.
While the mechanisms underlying the autoimmune phenomena and endocrinopathies associated with CMCC could be explained by defects in AIRE itself, the link between the IL-17 pathway and chronic candidiasis (which is frequently the presenting symptom and almost invariably present) was only recently elucidated.
The first clue came from reports of high titers of neutralizing antibodies to type I IFNs (Meager et al. 2006). In a study involving Finnish and Norwegian populations, almost all patients with APECED exhibited autoantibodies to IFN-α subtypes as well as IFN-ω. More recently, neutralizing antibodies against IL-17A, IL-17F, and IL-22 were also detected (Kisand et al. 2010; Puel et al. 2010). These cytokines are critical for the inflammatory response against Candida, and the presence of such autoantibodies likely explains candidiasis in these patients. Interestingly, susceptibility to candidal infection may result from both abnormal adaptive and innate immunity. Further studies are needed to fully decipher the pathophysiology of AIRE deficiency in CMCC, given the interaction of AIRE with the beta-glucan receptor Dectin-1 pathway components such as caspase recruitment domain-containing protein 9 (CARD9) and Syk (Pedroza et al. 2012). TNF-α production following stimulation of the Dectin-1 pathway is reduced in patients with AIRE deficiency. In addition, it appears that organ involvement and severity of disease is also affected by genetic background and modifying genes such as HLA (Betterle et al. 1998; Karvonen et al. 2000).
Signal transducer and activator of transcription-1 (STAT1) dysfunction
The signaling molecule STAT1 mediates cellular responses following type I and type II IFN receptor ligation, as well as IL-23 and IL-12 receptor engagement. Monoallelic mutations in STAT1 have been identified in patients with CMCC (van de Veerdonk et al. 2011), although the clinical manifestations were limited to the skin, nails, and mucous membranes, as well as increased incidence of hypothyroidism. The reported STAT1 gain-of-function mutations occurred in the coiled-coil domain, resulting in reduced production of IL-17, IL-22, and IFNγ, thereby linking these defects with aberrant Th17 cell activity. It was proposed that overexpression and activity of type I, type II, and type III IFNs inhibited Th17 differentiation (Liu et al. 2011; Maródi et al. 2012). However, this is in counter to recent findings highlighting the central role of IFNs in the protective host response to Candida (Smeekens et al. 2013a).
We recently described a group of CMCC patients with loss-of-function STAT1 mutations (Sharfe et al. 2014). These patients were phenotypically different from previous reports, where alongside chronic candidiasis, individuals also suffered recurrent infections with bacterial and viral pathogens (cytomegalovirus, John Cunningham virus) as well as severe autoimmune phenomena reminiscent of a combined immunodeficiency phenotype. Immunologic laboratory evaluation revealed progressive loss of T and B cell function.
It is important to note that mutations in STAT1 have been reported in several other phenotypically different immunodeficiencies, such as the Mendelian susceptibility to mycobacterial diseases (MSMD) (Boisson-Dupuis et al. 2012). A possible explanation for the diverse phenotypes include mutations which target different regions of STAT1, thereby affecting other cellular functions. Another explanation may be that defects in other modifying genes are present, resulting in different phenotypes.
Caspase recruitment domain-containing protein 9 (CARD9) deficiency
Mutations in the signal transduction molecule CARD9 have also been identified as a cause of CMCC. CARD9 signals downstream of Dectin-1 as well as Dectin-2 and Mincle (Gross et al. 2006; Robinson et al. 2009). The mode of inheritance appears to be autosomal dominant, with the phenotype consisting mainly of recurrent chronic candidiasis as well as systemic infections(Glocker et al. 2009; Lanternier et al. 2012).
Interleukin-17 pathway defects
Mutations in the cytokine IL-17F and cytokine receptor IL-17RA causing CMCC have been reported, highlighting the role of the IL-17 pathway in mucocutaneous immunity against Candida. One patient born to consanguineous parents presented with candida dermatitis, and was found to have a homozygous mutation in IL-17RA (Q248X) which was transmitted in an autosomal recessive manner. The patient’s peripheral mononuclear cells and fibroblasts lacked IL-17RA expression, resulting in absent responses to IL-17A/F (Puel et al. 2010). Levy and colleagues later reported a case series of 21 patients with IL-17RA mutations. While some had CMCC, others also suffered recurrent bacterial infections (Lévy et al. 2016). A separate kindred was reported to have a heterozygous mutation in IL-17F, showing impaired cytokine secretion (Puel et al. 2010). The inheritance was suggested to be autosomal dominant, although there were other asymptomatic family members with the same mutation, indicating a model of incomplete penetrance.
Biallelic mutations in ACT1, an adaptor protein downstream of the IL-17 receptor which activates nuclear factor κB (NFκB), MAPK, and C/EBP pathways, has been reported in a kindred of 2 brothers who presented with chronic oral candidiasis and recurrent blepharitis with S. aureus (Boisson et al. 2013). Mutations in IL-17RC, another member of the IL-17 pathway, resulted in IL-17RC deficiency in 3 unrelated kindreds involving an Argentinian and 2 Turkish patients. All had CMCC with no other infections, no autoimmune phenomena and a normal B and T cell immune workup (Ling et al. 2015). This same pathway is affected by biallelic mutations in RORC, resulting in deficiency of IL-17 producing T cells as well as defective INF-γ responses to mycobacteria. These patients from 3 kindreds presented with susceptibility to mycobacterial infections—this was fatal in 1 child with disseminated BCG infection. Most of the patients also had chronic candidiasis (Okada et al. 2015).
Signal transducer and activator of transcription-3 (STAT3) dysfunction
Some monogenic diseases may feature CMCC as a component of the clinical manifestations. In such cases, CMCC is not a predominant feature and may not always be present. Autosomal dominant hyper IgE syndrome (AD-HIES) has been shown to result in CMCC. The main clinical features include eczema, recurrent skin infections, cold abscesses, as well as recurrent severe pneumonia with pneumatocele formation, which is typical of S. aureus infections. Among these infections, persistent mucosal and skin candidiasis and other fungal infections are often seen. Patients may also have defects in bone formation resulting in skeletal defects, as well as coarse facial features.
AD-HIES is caused by mutations in STAT3 (Holland et al. 2007; Minegishi et al. 2007), a signaling molecule downstream of the IL-23 receptor. The defect in STAT3 results in reduction in Th17 cell numbers and absent IL-17 secretion (de Beaucoudrey et al. 2008).
Dedicator of cytokinesis 8 (DOCK8) deficiency
CMCC has been attributed to mutations in DOCK8, a member of the DOCK180-related protein family which is also involved in Th17 polarization (Engelhardt et al. 2009). The autosomal recessive form of HIES caused by defects in DOCK8 is phenotypically different, with patients showing highly elevated levels of IgE and eosinophilia. In this disorder, CMCC is not one of the main features but, rather, part of the severe combined immunodeficiency state with viral and bacterial infections (Zhang et al. 2009). Patients experience severe recurrent sinopulmonary infections, recurrent viral infections especially of the skin (herpes simplex, herpes zoster, molluscum contagiosum, and human papilloma virus), alongside atopic disease such as atopic dermatitis and food allergies. Hepatic disorders and high incidence of neoplastic disease, vulvar, facial, and anal squamous cell dysplasia and carcinomas, as well as T cell lymphoma-leukemia are also common. The overall immunologic profile of these patients, which include lymphopenia and reduced lymphocyte proliferation, is typical of combined immunodeficiency.
Tyrosine Kinase 2 (TYK2) deficiency
CMCC associated with HIES is caused by mutations in TYK2 (Minegishi et al. 2006), which encodes for a tyrosine kinase from the Janus kinase (JAK) family. The patient reported had characteristic features of both autosomal recessive HIES and atypical mycobacteriosis and chronic candidiasis.
CD25 deficiency
CD25 (or IL-2Rα) deficiency causes a combined immunodeficiency phenotype. Patients present with severe chronic candidiasis, bacterial and viral infections, as well as autoimmune phenomena (Sharfe et al. 1997). This autosomal recessive defect results in severe reduction in T cell numbers, with B cells showing normal development. Due to lack of regulatory mechanisms and abnormal proliferation, these patients experience lymphocytic infiltration of tissues, lung, liver, bone and skin causing inflammation and severe damage.
Patients with CD25 deficiency suffer from chronic candidiasis as well as invasive candidemia (Ouederni et al. 2014). Several genetic variants were also associated with CMCC, some with isolated candidiasis while others as part of a complex syndrome.
Protein tyrosine phosphatase non-receptor type-22 (PTPN22) mutations
Mutations in PTPN22, encoding Lyp, are associated with wide variety of autoimmune diseases. In particular, the R620W variant is found in significantly high prevalence in a group of CMCC patients (Nahum et al. 2008). The substrates for Lyp phosphatase activity include Lck, Fyn, Zap70 and the TCR zeta chain, and Lyp is also known to interact with Csk, an Src negative regulatory kinase. Whereas Lyp overexpression downregulates TCR signaling, Lyp deficiency has been shown to enhance T cell activation. The R620W variant is a gain-of-function mutation, resulting in defects in central as well as peripheral tolerance (Gregorieff et al. 1998; Gjörloff-Wingren et al. 1999; Vang et al. 2005), as seen in rheumatoid arthritis, systemic lupus erythromatosis, and adrenal insufficiency (Bottini et al. 2004; Wu et al. 2005; Michou et al. 2007). While the underlying mechanism is not completely understood, it may well be related to disruption of regulatory mechanisms which are known from other genetic defects to promote colonization with Candida.
Susceptibility genes associated with CMCC
Polymorphisms in genes that result in innate immune related defects are also responsible or associated with chronic candidiasis.
Such is the case of Dectin-1 single nucleotide polymorphisms (Ferwerda et al. 2009). Dectin-1 is an important sensor of Candida, which recognizes the microbial cell wall constituent β-glucan. The Y238X variant, found in as much as 8% of European and 40% of sub-Saharan African populations, has been suggested to confer susceptibility to mucosal candidiasis but not systemic candidemia (Rosentul et al. 2011). Reduced secretion of IL-1β and attenuation of Th17 responses have been reported for this variant (Plantinga et al. 2009).
Polymorphisms or variants in TLR receptors were also reported as susceptibility genes for chronic candidiasis. The TLR2 R753Q variant was shown in 1 study to be associated with candidiasis through reduced IFN-γ and IL-8 secretion (Woehrle et al. 2008). The TLR4 variants, D299G and Y399I, have also been reported as risk factors for invasive blood stream candidemia (Van der Graaf et al. 2006). We have previously shown that a variant of TLR3, L412F, confers susceptibility to CMCC (Nahum et al. 2011). Responses to stimulation with Candida as well as TLR3 ligands were significantly abrogated, resulting in reduced secretion IFN-γ (Nahum et al. 2012).
Recently, polymorphisms in TLR1 were shown to contribute to high risk of candidemia, likely through effects on IL-8 and IFN-γ levels (Plantinga et al. 2012). Recurrent vulvo-vaginal candidiasis is a common finding in individuals with polymorphisms in the inflammasome protein NLRP3 (Lev-Sagie et al. 2009), as well another innate receptor gene, MBL2, which encodes for mannose binding lectin (Babula et al. 2003; Giraldo et al. 2007). Recent studies suggest an important role for the inflammasome product IL-1β in eliciting a proper T cell mediated responses to Candida (Zielinski et al. 2012).
Chronic disseminated candidiasis has also been reported in association with changes in IL-4 (Choi et al. 2003), and persistent candidemia shown with variants of IL-12b and IL-10 (Johnson et al. 2012).
Concluding remarks
Chronic candidiasis is a feature present in a broad range of diseases, from isolated candidiasis involving skin and mucous membrane, to syndromes encompassing severe immunodeficiency and autoimmunity. A careful immune workup is therefore necessary, and should include thorough genetic and functional studies. Issues to consider include different treatment modalities (as is the case for STAT1 loss-of-function mutations, where bone marrow transplantation may be of benefit but is complicated by the many phenotypes associated with this gene) and the possibility that prolonged and repeated courses of anti-fungal treatment may result in microbial resistance.
It is important to note that although monogenic immunodeficiency disorders are interesting and insightful with regard to mechanisms of infection; in the vast majority of Candida infections, the cause is multifactorial. These include susceptibility genes found in large numbers of the general population in combination with factors such as altered local microbial flora (resulting from use of antibiotics, immunomodulators, and chemotherapy).
Together, these stress the need for improved understanding of the mechanisms underlying susceptibility to Candida infection, which may pave the way for new and innovative treatment strategies. Of equal importance is the implementation of large population-based genetic studies combined with methods from the fields of functional genomics and mathematical-biological methods. Targeting future treatments towards key molecules may prove to be beneficial to a large number of patients suffering from Candida infections such as those with hematological malignancies and other immunocompromised patients.
REFERENCES
Ahonen P., Myllärniemi S., Sipilä I., and Perheentupa J. 1990. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 322(26):1829–1836.
Akira S., Uematsu S., and Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell. 124(4):783–801.
Babula O., Lazdane G., Kroica J., Ledger W.J., and Witkin S.S. 2003. Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin. Infect. Dis. 37(5):733–737.
Betterle C., Greggio N.A., and Volpato M. 1998. Clinical review 93: Autoimmune polyglandular syndrome type 1. J. Clin. Endocrinol. Metab. 83(4):1049–1055.
Björses P., Aaltonen J., Horelli-Kuitunen N., Yaspo M.L., and Peltonen L. 1998. Gene defect behind APECED: A new clue to autoimmunity. Hum. Mol. Genet. 7(10):1547–1553.
Boisson B., Wang C., Pedergnana V., Wu L., Cypowyj S., Rybojad M., Belkadi A., Picard C., Abel L., Fieschi C., Puel A., Li X., and Casanova J.L. 2013. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 39(4):676–686.
Boisson-Dupuis S., Kong X.-F., Okada S., Cypowyj S., Puel A., Abel L., and Casanova J.L. 2012. Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 24(4):364–378.
Bottini N., Musumeci L., Alonso A., Rahmouni S., Nika K., Rostamkhani M., MacMurray J., Meloni G.F., Lucarelli P., Pellecchia M., Eisenbarth G.S., Comings D., and Mustelin T. 2004. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 36(4):337–338.
Brown G.D. and Gordon S. 2001. Immune recognition: A new receptor for β-glucans. Nature. 413(6851):36–37.
Chitnis A.S., Magill S.S., Edwards J.R., Chiller T.M., Fridkin S.K., and Lessa F.C. 2012. Trends in Candida central line-associated bloodstream infections among NICUs, 1999–2009. Pediatrics. 130(1):e46–e52.
Choi E.H., Foster C.B., Taylor J.G., Erichsen H.C., Chen R.A., Walsh T.J., Anttila V.J., Ruutu T., Palotie A., and Chanock S.J. 2003. Association between chronic disseminated candidiasis in adult acute leukemia and common IL4 promoter haplotypes. J. Infect. Dis. 187(7):1153–1156.
Das I., Nightingale P., Patel M., and Jumaa P. 2011. Epidemiology, clinical characteristics, and outcome of candidemia: Experience in a tertiary referral center in the UK. Int. J. Infect. Dis. 15(11):e759–e763.
de Beaucoudrey L., Puel A., Filipe-Santos O., Cobat A., Ghandil P., Chrabieh M., Feinberg J., von Bernuth H., Samarina A., Jannière L., Fieschi C., Stéphan J.L., Boileau C., Lyonnet S., Jondeau G., Cormier-Daire V., Le Merrer M., Hoarau C., Lebranchu Y., Lortholary O., Chandesris M.O., Tron F., Gambineri E., Bianchi L., Rodriguez-Gallego C., Zitnik S.E., Vasconcelos J., Guedes M., Vitor A.B., Marodi L., Chapel H., Reid B., Roifman C., Nadal D., Reichenbach J., Caragol I., Garty B.Z., Dogu F., Camcioglu Y., Gülle S., Sanal O., Fischer A., Abel L., Stockinger B., Picard C., and Casanova J.L. 2008. Mutations in STAT3 and IL12RB1 impair the development of human IL-17–producing T cells. J. Exp. Med. 205(7):1543–1550.
Donlan R.M. and Costerton J.W. 2002. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15(2):167–193.
Engelhardt K.R., McGhee S., Winkler S., Sassi A., Woellner C., Lopez-Herrera G., Chen A., Kim H.S., Lloret M.G., Schulze I., Ehl S., Thiel J., Pfeifer D., Veelken H., Niehues T., Siepermann K., Weinspach S., Reisli I., Keles S., Genel F., Kutukculer N., Camcioğlu Y., Somer A., Karakoc-Aydiner E., Barlan I., Gennery A., Metin A., Degerliyurt A., Pietrogrande M.C., Yeganeh M., Baz Z., Al-Tamemi S., Klein C., Puck J.M., Holland S.M., McCabe E.R., Grimbacher B., and Chatila T.A. 2009. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J. Allergy Clin. Immunol. 124(6):1289–1302.e4.
Ferwerda B., Ferwerda G., Plantinga T.S., Willment J.A., van Spriel A.B., Venselaar H., Elbers C.C., Johnson M.D., Cambi A., Huysamen C., Jacobs L., Jansen T., Verheijen K., Masthoff L., Morré S.A., Vriend G., Williams D.L., Perfect J.R., Joosten L.A., Wijmenga C., van der Meer J.W., Adema G.J., Kullberg B.J., Brown G.D., and Netea M.G. 2009. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 361(18):1760–1767.
Giraldo P.C., Babula O., Gonçalves A.K.S., Linhares I.M., Amaral R.L., Ledger W.J., and Witkin S.S. 2007. Mannose-binding lectin gene polymorphism, vulvovaginal candidiasis, and bacterial vaginosis. Obstet. Gynecol. 109(5):1123–1128.
Gjörloff-Wingren A., Saxena M., Williams S., Hammi D., and Mustelin T. 1999. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur. J. Immunol. 29(12):3845–3854.
Glocker E.-O., Hennigs A., Nabavi M., Schäffer A.A., Woellner C., Salzer U., Pfeifer D., Veelken H., Warnatz K., Tahami F., Jamal S., Manguiat A., Rezaei N., Amirzargar A.A., Plebani A., Hannesschläger N., Gross O., Ruland J., and Grimbacher B. 2009. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 361(18):1727–1735.
Gregorieff A., Cloutier J.F., and Veillette A. 1998. Sequence requirements for association of protein-tyrosine phosphatase PEP with the Src homology 3 domain of inhibitory tyrosine protein kinase p50(csk). J. Biol. Chem. 273(21):13217–13222.
Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., and Ruland J. 2006. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 442(7103):651–656.
Hernández-Santos N. and Gaffen S.L. 2012. Th17 cells in immunity to Candida albicans. Cell Host Microbe. 11(5):425–435.
Holland S.M., DeLeo F.R., Elloumi H.Z., Hsu A.P., Uzel G., Brodsky N., Freeman A.F., Demidowich A., Davis J., Turner M.L., Anderson V.L., Darnell D.N., Welch P.A., Kuhns D.B., Frucht D.M., Malech H.L., Gallin J.I., Kobayashi S.D., Whitney A.R., Voyich J.M., Musser J.M., Woellner C., Schäffer A.A., Puck J.M., and Grimbacher B. 2007. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 357(16):1608–1619.
Ivanov I.I., McKenzie B.S., Zhou L., Tadokoro C.E., Lepelley A., Lafaille J.J., Cua D.J., and Littman D.R. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 126(6):1121–1133.
Iwakura Y., Ishigame H., Saijo S., and Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity. 34(2):149–62.
Johnson M.D., Plantinga T.S., van de Vosse E., Velez Edwards D.R., Smith P.B., Alexander B.D., Yang J.C., Kremer D., Laird G.M., Oosting M., Joosten L.A., van der Meer J.W., van Dissel J.T., Walsh T.J., Perfect J.R., Kullberg B.J., Scott W.K., and Netea M.G. 2012. Cytokine gene polymorphisms and the outcome of invasive candidiasis: A prospective cohort study. Clin. Infect. Dis. 54(4):502–510.
Jouault T., Ibata-Ombetta S., Takeuchi O., Trinel P., Sacchetti P., Lefebvre P., Akira S., and Poulain D. 2003. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 188(1):165–172.
Karvonen M., Viik-Kajander M., Moltchanova E., Libman I., LaPorte R., and Tuomilehto J. 2000. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) Project Group. Diabetes Care. 23(10):1516–1526.
Kirkpatrick C.H. 1994. Chronic mucocutaneous candidiasis. J. Am. Acad. Dermatol. 31(3):S14–S17.
Kisand K., Bøe Wolff A.S., Podkrajšek K.T., Tserel L., Link M., Kisand K.V., Ersvaer E., Perheentupa J., Erichsen M.M., Bratanic N., Meloni A., Cetani F., Perniola R., Ergun-Longmire B., Maclaren N., Krohn K.J., Pura M., Schalke B., Ströbel P., Leite M.I., Battelino T., Husebye E.S., Peterson P., Willcox N., and Meager A. 2010. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207(2):299–308.
Lanternier F., Pathan S., Vincent Q., Liu L., Cypowij S., Prando C., Migaud M., Taibi L., Ammar-Khodja A., Stambouli O.B., Guellil B., Jacobs F., Goffard J.-C., Shepers K., del Marmol V., Bachelez H., Michel L., Lefranc G., Fraitag S., Bougnoux M.-E., and Boudia A. 2012. Human invasive dermatophytic disease is caused by inborn errors of CARD9. J. Clin. Immunol. 32:1–379.
Lev-Sagie A., Prus D., Linhares I.M., Lavy Y., Ledger W.J., and Witkin S.S. 2009. Polymorphism in a gene coding for the inflammasome component NALP3 and recurrent vulvovaginal candidiasis in women with vulvar vestibulitis syndrome. Am. J. Obstet. Gynecol. 200(3):303.e1–303.e6.
Lévy R., Okada S., Béziat V., Moriya K., Liu C., Chai L.Y.A., Migaud M., Hauck F., Al Ali A., Cyrus C., Vatte C., Patiroglu T., Unal E., Ferneiny M., Hyakuna N., Nepesov S., Oleastro M., Ikinciogullari A., Dogu F., Asano T., Ohara O., Yun L., Della Mina E., Bronnimann D., Itan Y., Gothe F., Bustamante J., Boisson-Dupuis S., Tahuil N., Aytekin C., Salhi A., Al Muhsen S., Kobayashi M., Toubiana J., Abel L., Li X., Camcioglu Y., Celmeli F., Klein C., AlKhater S.A., Casanova J.L., and Puel A. 2016. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc. Natl. Acad. Sci. USA. 113(51):E8277–E8285.
Ling Y., Cypowyj S., Aytekin C., Galicchio M., Camcioglu Y., Nepesov S., Ikinciogullari A., Dogu F., Belkadi A., Levy R., Migaud M., Boisson B., Bolze A., Itan Y., Goudin N., Cottineau J., Picard C., Abel L., Bustamante J., Casanova J.L., and Puel A. 2015. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 212(5):619–631.
Liu L., Okada S., Kong X.-F., Kreins A.Y., Cypowyj S., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P., Masson C., Toth B., Flatot J., Migaud M., Chrabieh M., Kochetkov T., Bolze A., Borghesi A., Toulon A., Hiller J., Eyerich S., Eyerich K., Gulácsy V., Chernyshova L., Chernyshov V., Bondarenko A., Grimaldo R.M., Blancas-Galicia L., Beas I.M., Roesler J., Magdorf K., Engelhard D., Thumerelle C., Burgel P.R., Hoernes M., Drexel B., Seger R., Kusuma T., Jansson A.F., Sawalle-Belohradsky J., Belohradsky B., Jouanguy E., Bustamante J., Bué M., Karin N., Wildbaum G., Bodemer C., Lortholary O., Fischer A., Blanche S., Al-Muhsen S., Reichenbach J., Kobayashi M., Rosales F.E., Lozano C.T., Kilic S.S., Oleastro M., Etzioni A., Traidl-Hoffmann C., Renner E.D., Abel L., Picard C., Maródi L., Boisson-Dupuis S., Puel A., and Casanova J.L. 2011. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208(8):1635–1648.
Maródi L., Cypowyj S., Tóth B., Chernyshova L., Puel A., and Casanova J.L. 2012. Molecular mechanisms of mucocutaneous immunity against Candida and Staphylococcus species. J. Allergy Clin. Immunol. 130(5):1019–1027.
McGeachy M.J. and McSorley S.J. 2012. Microbial-induced Th17: Superhero or supervillain? J Immunol. 189(7):3285–3291.
McGreal E.P., Rosas M., Brown G.D., Zamze S., Wong S.Y.C., Gordon S., Martinez-Pomares L., and Taylor P.R. 2006. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology. 16(5):422–430.
Meager A., Visvalingam K., Peterson P., Möll K., Murumägi A., Krohn K., Eskelin P., Perheentupa J., Husebye E., Kadota Y., Willcox N., and Willcox N. 2006. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 3(7):e289.
Michou L., Lasbleiz S., Rat A.-C., Migliorini P., Balsa A., Westhovens R., Barrera P., Alves H., Pierlot C., Glikmans E., Garnier S., Dausset J., Vaz C., Fernandes M., Petit-Teixeira E., Lemaire I., Pascual-Salcedo D., Bombardieri S., Dequeker J., Radstake T.R., Van Riel P., van de Putte L., Lopes-Vaz A., Prum B., Bardin T., Dieudé P., and Cornélis F., andEuropean Consortium on Rheumatoid Arthritis Families. 2007. Linkage proof for PTPN22, a rheumatoid arthritis susceptibility gene and a human autoimmunity gene. Proc. Natl. Acad. Sci. USA. 104(5):1649–1654.
Minegishi Y., Saito M., Morio T., Watanabe K., Agematsu K., Tsuchiya S., Takada H., Hara T., Kawamura N., Ariga T., Kaneko H., Kondo N., Tsuge I., Yachie A., Sakiyama Y., Iwata T., Bessho F., Ohishi T., Joh K., Imai K., Kogawa K., Shinohara M., Fujieda M., Wakiguchi H., Pasic S., Abinun M., Ochs H.D., Renner E.D., Jansson A., Belohradsky B.H., Metin A., Shimizu N., Mizutani S., Miyawaki T., Nonoyama S., Karasuyama H., and Karasuyama H. 2006. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 25(5):745–755.
Minegishi Y., Saito M., Tsuchiya S., Tsuge I., Takada H., Hara T., Kawamura N., Ariga T., Pasic S., Stojkovic O., Metin A., and Karasuyama H. 2007. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 448(7157):1058–1062.
Nagamine K., Peterson P., Scott H.S., Kudoh J., Minoshima S., Heino M., Krohn K.J., Lalioti M.D., Mullis P.E., Antonarakis S.E., Kawasaki K., Asakawa S., Ito F., and Shimizu N. 1997. Positional cloning of the APECED gene. Nat. Genet. 17(4):393–398.
Nahum A., Bates A., Sharfe N., and Roifman C.M. 2008. Association of the lymphoid protein tyrosine phosphatase, R620W variant, with chronic mucocutaneous candidiasis. J. Allergy Clin. Immunol. 122(6):1220–1222.
Nahum A., Dadi H., Bates A., and Roifman C.M. 2011. The L412F variant of Toll-like receptor 3 (TLR3) is associated with cutaneous candidiasis, increased susceptibility to cytomegalovirus, and autoimmunity. J. Allergy Clin. Immunol. 127(2):528–531.
Nahum A., Dadi H., Bates A., and Roifman C.M. 2012. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun. Rev. 11(5):341–347.
Netea M.G., Gow N.A.R., Munro C.A., Bates S., Collins C., Ferwerda G., Hobson R.P., Bertram G., Hughes H.B., Jansen T., Jacobs L., Buurman E.T., Gijzen K., Williams D.L., Torensma R., McKinnon A., MacCallum D.M., Odds F.C., Van der Meer J.W., Brown A.J., and Kullberg B.J. 2006. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J. Clin. Invest. 116(6): 1642–1650.
Okada S., Markle J.G., Deenick E.K., Mele F., Averbuch D., Lagos M., Alzahrani M., Al-Muhsen S., Halwani R., Ma C.S., Wong N., Soudais C., Henderson L.A., Marzouqa H., Shamma J., Gonzalez M., Martinez-Barricarte R., Okada C., Avery D.T., Latorre D., Deswarte C., Jabot-Hanin F., Torrado E., Fountain J., Belkadi A., Itan Y., Boisson B., Migaud M., Arlehamn C.S.L., Sette A., Breton S., McCluskey J., Rossjohn J., de Villartay J.P., Moshous D., Hambleton S., Latour S., Arkwright P.D., Picard C., Lantz O., Engelhard D., Kobayashi M., Abel L., Cooper A.M., Notarangelo L.D., Boisson-Dupuis S., Puel A., Sallusto F., Bustamante J., Tangye S.G., and Casanova J.L. 2015. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 349(6248):606–613.
Ortega M., Rovira M., Almela M., Marco F., de la Bellacasa J.P., Martínez J.A., Carreras E., and Mensa J. 2005. Bacterial and fungal bloodstream isolates from 796 hematopoietic stem cell transplant recipients between 1991 and 2000. Ann. Hematol. 84(1):40–46.
Ouederni M., Sanal O., Ikinciogullari A., Tezcan I., Dogu F., Sologuren I., Pedraza-Sánchez S., Keser M., Tanir G., Nieuwhof C., Colino E., Kumararatne D., Levy J., Kutukculer N., Aytekin C., Herrera-Ramos E., Bhatti M., Karaca N., Barbouche R., Broides A., Goudouris E., Franco J.L., Parvaneh N., Reisli I., Strickler A., Shcherbina A., Somer A., Segal A., Angel-Moreno A., Lezana-Fernandez J.L., Bejaoui M., Bobadilla-Del Valle M., Kachboura S., Sentongo T., Ben-Mustapha I., Bustamante J., Picard C., Puel A., Boisson-Dupuis S., Abel L., Casanova J.L., and Rodríguez-Gallego C. 2014. Clinical features of Candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin. Infect. Dis. 58(2):204–213.
Pedroza L.A., Kumar V., Sanborn K.B., Mace E.M., Niinikoski H., Nadeau K., Vasconcelos Dde M., Perez E., Jyonouchi S., Jyonouchi H., Banerjee P.P., Ruuskanen O., Condino-Neto A., and Orange J.S. 2012. Autoimmune regulator (AIRE) contributes to Dectin-1-induced TNF-α production and complexes with caspase recruitment domain-containing protein 9 (CARD9), spleen tyrosine kinase (Syk), and Dectin-1. J. Allergy Clin. Immunol. 129(2):464–472, 472.e1–472.e3.
Perheentupa J. 2006. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 91(8):2843–2850.
Plantinga T.S., Johnson M.D., Scott W.K., van de Vosse E., Velez Edwards D.R., Smith P.B., Alexander B.D., Yang J.C., Kremer D., Laird G.M., Oosting M., Joosten L.A., van der Meer J.W., van Dissel J.T., Walsh T.J., Perfect J.R., Kullberg B.J., and Netea M.G. 2012. Toll-like receptor 1 polymorphisms increase susceptibility to candidemia. J. Infect. Dis. 205(6):934–943.
Plantinga T.S., van der Velden W.J.F.M., Ferwerda B., van Spriel A.B., Adema G., Feuth T., Donnelly J.P., Brown G.D., Kullberg B.J., Blijlevens N.M., and Netea M.G. 2009. Early stop polymorphism in human DECTIN-1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 49(5):724–732.
Puel A., Cypowyj S., Bustamante J., Wright J.F., Liu L., Lim H.K., Migaud M., Israel L., Chrabieh M., Audry M., Gumbleton M., Toulon A., Bodemer C., El-Baghdadi J., Whitters M., Paradis T., Brooks J., Collins M., Wolfman N.M., Al-Muhsen S., Galicchio M., Abel L., Picard C., and Casanova J.L. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 332(6025):65–68.
Puel A., Döffinger R., Natividad A., Chrabieh M., Barcenas-Morales G., Picard C., Cobat A., Ouachée-Chardin M., Toulon A., Bustamante J., Al-Muhsen S., Al-Owain M., Arkwright P.D., Costigan C., McConnell V., Cant A.J., Abinun M., Polak M., Bougnères P.F., Kumararatne D., Marodi L., Nahum A., Roifman C., Blanche S., Fischer A., Bodemer C., Abel L., Lilic D., and Casanova J.L. 2010. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 207(2):291–297.
Robinson M.J., Osorio F., Rosas M., Freitas R.P., Schweighoffer E., Gross O., Verbeek J.S., Ruland J., Tybulewicz V., Brown G.D., Moita L.F., Taylor P.R., and Reis e Sousa C. 2009. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 206(9):2037–2051.
Rogers N.C., Slack E.C., Edwards A.D., Nolte M.A., Schulz O., Schweighoffer E., Williams D.L., Gordon S., Tybulewicz V.L., Brown G.D., and Reis e Sousa C. 2005. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity. 22(4):507–517.
Rosentul D.C., Plantinga T.S., Oosting M., Scott W.K., Velez Edwards D.R., Smith P.B., Alexander B.D., Yang J.C., Laird G.M., Joosten L.A., van der Meer J.W., Perfect J.R., Kullberg B.J., Netea M.G., and Johnson M.D. 2011. Genetic variation in the Dectin-1/CARD9 recognition pathway and susceptibility to candidemia. J. Infect. Dis. 204(7):1138–1145.
Rutz S., Eidenschenk C., and Ouyang W. 2013. IL-22, not simply a Th17 cytokine. Immunol. Rev. 252(1):116–132.
Sharfe N., Dadi H.K., Shahar M., and Roifman C.M. 1997. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA. 94(7):3168–3171.
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., Herbrick J.A., and Roifman C.M. 2014. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J. Allergy. Clin. Immunoly. 133(3):807–817.
Smeekens S.P., Ng A., Kumar V., Johnson M.D., Plantinga T.S., van Diemen C., Arts P., Verwiel E.T., Gresnigt M.S., Fransen K., van Sommeren S., Oosting M., Cheng S.C., Joosten L.A., Hoischen A., Kullberg B.J., Scott W.K., Perfect J.R., van der Meer J.W., Wijmenga C., Netea M.G., and Xavier R.J. 2013a. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat. Commun. 4:1342.
Smeekens S.P., van de Veerdonk F.L., Kullberg B.J., and Netea M.G. 2013b. Genetic susceptibility to Candida infections. EMBO Mol. Med. 5(6):805–813.
Sobel J.D. 2007. Vulvovaginal candidosis. Lancet. 369(9577):1961–1971.
Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E., Bonilla F.A., Pechacek J., Chandrasekaran P., Browne S.K., Agharahimi A., Gharib A.M., Mannurita S.C., Yim J.J., Gambineri E., Torgerson T., Tran D.Q., Milner J.D., and Holland S.M. 2013. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation–polyendocrinopathy–enteropathy–X-linked–like syndrome. J. Allergy Clin. Immunol. 131(6):1611–1623.
van de Veerdonk F.L., Plantinga T.S., Hoischen A., Smeekens S.P., Joosten L.A., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A., Kullberg B.J., van der Meer J.W., Lilic D., Veltman J.A., and Netea M.G. 2011. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365(1):54–61.
Van der Graaf C.A.A., Netea M.G., Morré S.A., Den Heijer M., Verweij P.E., Van der Meer J.W.M., and Kullberg B.J. 2006. Toll-like receptor 4 Asp299Gly/Thr399Ile polymorphisms are a risk factor for Candida bloodstream infection. Eur. Cytokine Netw. 17(1):29–34.
Vang T., Congia M., Macis M.D., Musumeci L., Orrú V., Zavattari P., Nika K., Tautz L., Taskén K., Cucca F., Mustelin T., and Bottini N. 2005. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat. Genet. 37(12):1317–1319.
Viscoli C., Girmenia C., Marinus A., Collette L., Martino P., Vandercam B., Doyen C., Lebeau B., Spence D., Krcmery V., De Pauw B., and Meunier F. 1999. Candidemia in cancer patients: A prospective, multicenter surveillance study by the Invasive Fungal Infection Group (IFIG) of the European Organization for Research and Treatment of Cancer (EORTC). Clin. Infect. Dis. 28(5):1071–1079.
Woehrle T., Du W., Goetz A., Hsu H.-Y., Joos T.O., Weiss M., Bauer U., Brueckner U.B., and Marion Schneider E. 2008. Pathogen specific cytokine release reveals an effect of TLR2 Arg753Gln during Candida sepsis in humans. Cytokine. 41(3):322–329.
Wu H., Cantor R.M., Graham D.S.C., Lingren C.M., Farwell L., Jager P.L., Bottini N., Grossman J.M., Wallace D.J., Hahn B.H., Julkunen H., Hebert L.A., Rovin B.H., Birmingham D.J., Rioux J.D., Yu C.Y., Kere J., Vyse T.J., and Tsao B.P. 2005. Association analysis of the R620W polymorphism of protein tyrosine phosphatase PTPN22 in systemic lupus erythematosus families: Increased T allele frequency in systemic lupus erythematosus patients with autoimmune thyroid disease. Arthritis. Rheum. 52(8):2396–2402.
Yang X.O., Panopoulos A.D., Nurieva R., Chang S.H., Wang D., Watowich S.S., and Dong C. 2007. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282(13):9358–9363.
Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G., Holland S.M., and Su H.C. 2009. Combined immunodeficiency associated with DOCK8 mutations. N. Engl. J. Med. 361(21):2046–2055.
Zielinski C.E., Mele F., Aschenbrenner D., Jarrossay D., Ronchi F., Gattorno M., Monticelli S., and Lanzavecchia A Sallusto F. 2012. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature. 484(7395): 514–518.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 3 • September 2017
Pages: 87 - 99
History
Received: 4 August 2017
Accepted: 5 September 2017
Accepted manuscript online: 7 September 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AmitNahum. 2017. Chronic mucocutaneous candidiasis: a spectrum of genetic disorders. LymphoSign Journal.
4(3): 87-99. https://doi.org/10.14785/lymphosign-2017-0011
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Chronic mucocutaneous candidiasis associated with a novel frameshift mutation in IL-17 receptor alpha
2. Abstracts from the Immunodeficiency Canada—6th SCID Symposium, Halifax, NS, 13 September 2018
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
