We describe an early-onset FHL-2 case showing the novel MACPF homozygous missense mutation c.917G>A which leads to the amino acid change p.Gly306Asp. Different amino acid substitutions that are located at the same position (c.916G>T leading to p.Gly306Cys (
Grossman et al. 2005;
Abdalgani et al. 2015), c.916G>A leading to p.Gly306Ser (
Nagafuji et al. 2007;
Wang et al. 2012;
Gao et al. 2016)) or neighbouring glycine (c.914C>A, leading to p.Gly305Asp, (
Clementi et al. 2005)) have been previously reported in very heterogeneous FHL-2 patients. We performed perforin immunophenotyping and
in silico analysis to ascertain the relevance of the novel p.Gly306Asp as well as the above mentioned mutations in HLH pathogenesis.
Functional and clinical presentation
Case report: A 19-month-old girl, the first child of a consanguineous couple with no previous familial history of hemophagocytic lympohistiocytosis (HLH), was first admitted to hospital because of fever due to unknown origin. Progressive anemia (haemoglobin 7.9 g/L), thrombopenia (8000/μl), hypertrigliceridemia (720 mg/dL), elevated ferritin (3373 ng/mL), hypofibrinoginemia (120 mg/dL) and hepatosplenomegaly, along with hemophagocytosis in bone marrow led to diagnosis of HLH, and she was started on the HLH-2004 therapeutic protocol (
Henter et al. 2007). According to the immunological results, she was classified as FHL-2 and after HLH-2004 induction therapy and conditioning, she underwent cord blood HCT (1.36×10
5 CD34
+ cells). Continuing maintenance treatment was required post-HCT and the patient unfortunately died at 150 days post-transplant because of graft failure and HLH disease reactivation.
Perforin expression was assessed by flow cytometry using anti-T cell receptor FITC, anti-CD8 PerCP, and anti-CD56 APC (BD Biosciences, San Jose, CA) antibodies for surface staining and anti-perforin PE (clone DG9, Pharmingen, BD Bioscience) or anti-granzyme B PE (Immunotech) antibodies for intracellular staining, as previously described (
Urrea Moreno et al. 2009). Samples were acquired in a FACSCalibur cytometer and analysed with FlowJo 7.6.1 software.
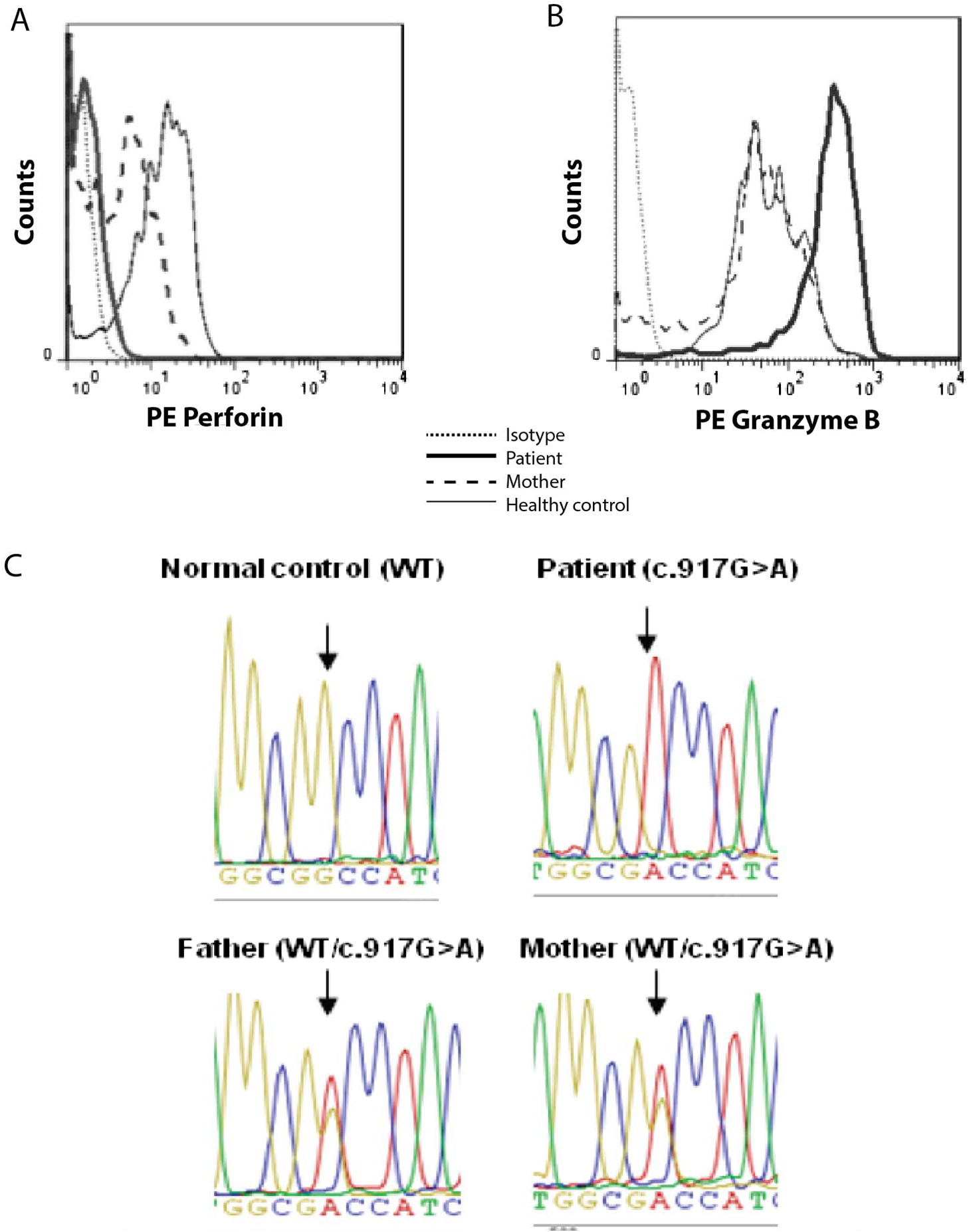
Figure 1A shows that perforin staining within patient’s circulating NK lymphocytes (corresponding to 16% of total lymphocytes) was as negative as the fluorescence intensity of isotypic control. On the other hand, granzyme-B expression—analyzed as a positive intracellular staining—was increased in the patient when compared to the patient´s mother and a healthy individual tested the same day of the analysis (
Figure 1B).
The mutational analysis was performed on genomic DNA from peripheral blood. Coding region of
PRF1 (exons 2 and 3) was amplified by PCR (Gene Amp PCR System 9700, PE Applied Biosystems, Warrington, UK) and sequenced (ABI PRISM 310 Genetic Analyzer from Applied Biosystems) as described (
Urrea Moreno et al. 2009). Chromatograms were created with Geneious Pro 5.5.7 and sequences were compared with reference
PRF1 (RefSeq: NG_009615.1).
Figure 1C shows the homozygous mutation c. 917G>A in exon 3 of
PRF1 that was found in the patient; this nucleotide change results in the non-conservative substitution of the non-polar amino acid glycine at position 306 of wild-type perforin protein with the negatively charged aspartic acid (p.Gly306Asp) in MACPF domain. The parents were both found to be heterozygous carriers of the mutated allele (
Figure 1C), correlating with the decreased perforin expression found in the mother (
Figure 1A).
Figure 2 shows a molecular model of perforin and of perfringolysin (PFO), a bacterial cholesterol dependant cytolysin (CDC), to illustrate the structural homology of their 2 pore-forming domains. 3D models were performed with Yasara View, v16.7.22 (
http://yasara.org/) (
Krieger and Vriend 2014). Gly306 and the preceding Gly305 are located at the beginning of a helical loop region that connects β4 and β4′ strands of the central beta sheet in both MACPFs and CDCs (
Lukoyanova et al. 2016;
Dudkina et al. 2016). As shown in
Figure 2, no proximity exists between the helical loop in perforin and the putative residues involved in its oligomerization, as opposed to what has been previously described in PFO (
Ramachandran et al. 2004).
For amino acid conservation analysis, representative sequences were obtained from the NCBI database (
www.ncbi.nlm.nih.gov). Alignments were performed with PROMALS3D (
http://prodata.swmed.edu/promals3d/promals3d.php), and edited with Geneious Pro 5.5.7 using Blosum45 matrix.
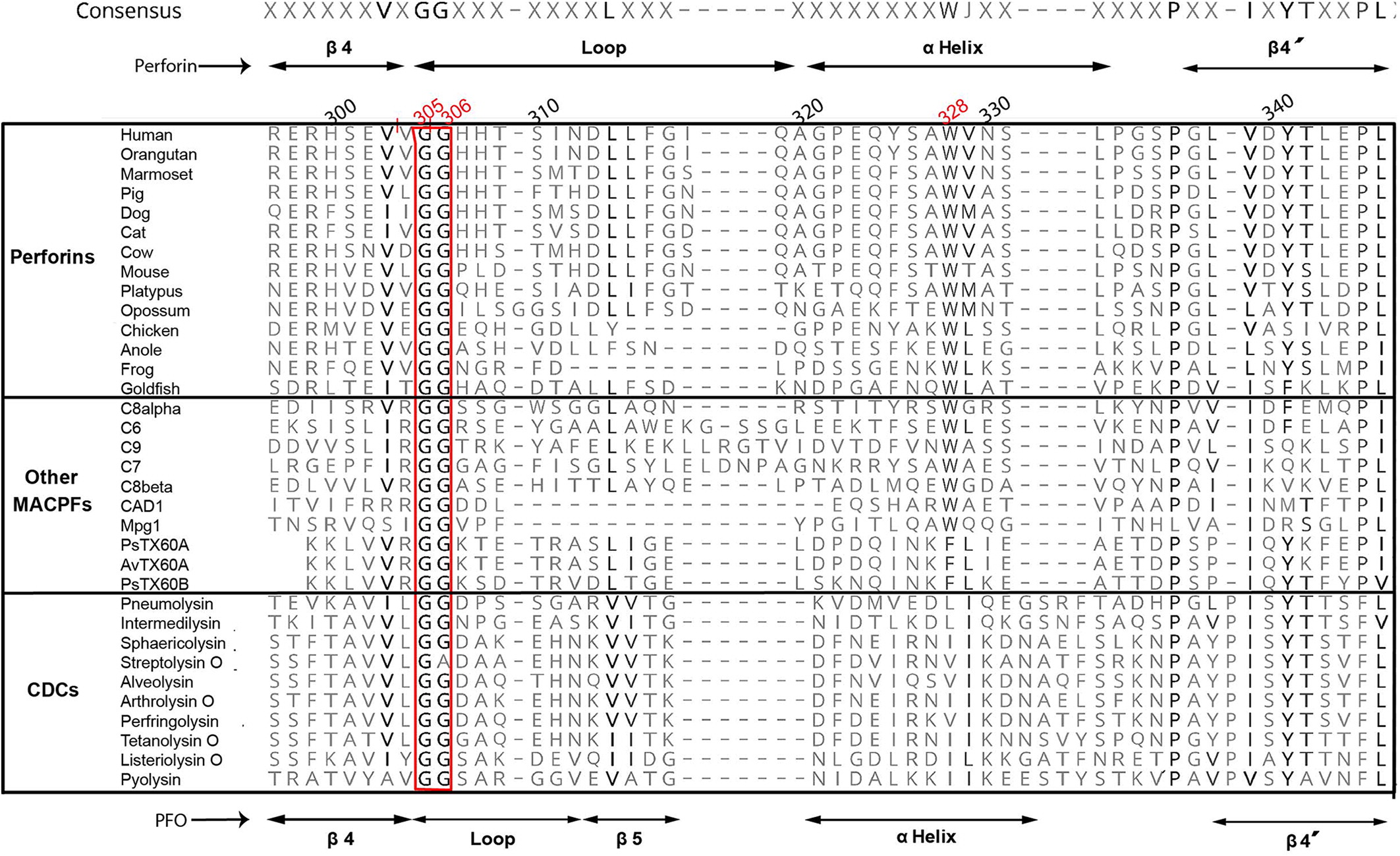
Figure 3 shows that both Gly305 and Gly306 in human perforin molecule or corresponding glycines in other vertebrate perforins, as well as other MACPF domains from different proteins, or even in pore-forming domains of bacterial cholesterol CDCs, constitute a highly conserved motif in all these proteins despite the low global sequence homology.
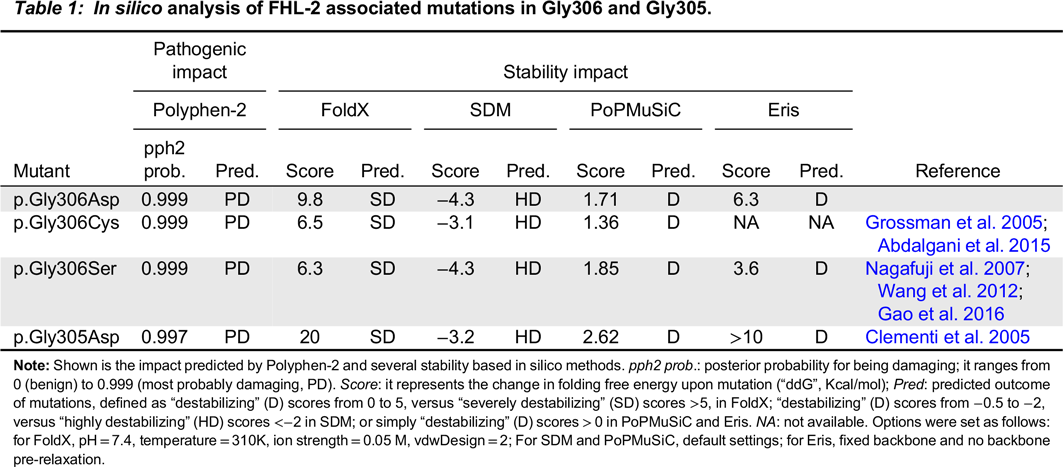
We next studied the impact of the above mutations on perforin thermodynamic stability, or the change in folding free energy upon mutation (i.e., differences in Gibb’s free energy between the folded and unfolded state of mutant compared to wild-type molecules, also ΔΔG or ddG). For this, we generated a molecular model of human perforin, using BuildModel and RepairPDB commands within FoldX v4 (
http://foldxsuite.crg.eu) (
Guerois et al. 2002), employing perforin X-ray crystal structure (PDB identifier:3nsj) (
Law et al. 2010) as a template. Independent algorithms used for stability calculations were FoldX itself, SDM (
http://mordred.bioc.cam.ac.uk/∼sdm/sdm.php) (
Worth et al. 2011), PoPMuSiC (
http://dezyme.com/) (
Dehouck et al. 2009) and Eris (
http://troll.med.unc.edu/eris) (
Yin et al. 2007). In support of our hypothesis, all 4 algorithms predicted that p.Gly306Asp, as well as the other 3 FHL-2 mutations reported in these glycines, i.e., p.Gly305Asp, p.Gly306Cys and p.Gly306Ser result in severe destabilization of the perforin molecule (
Table 1), albeit to a lesser extent in p.Gly306Ser according to Eris prediction. As shown in
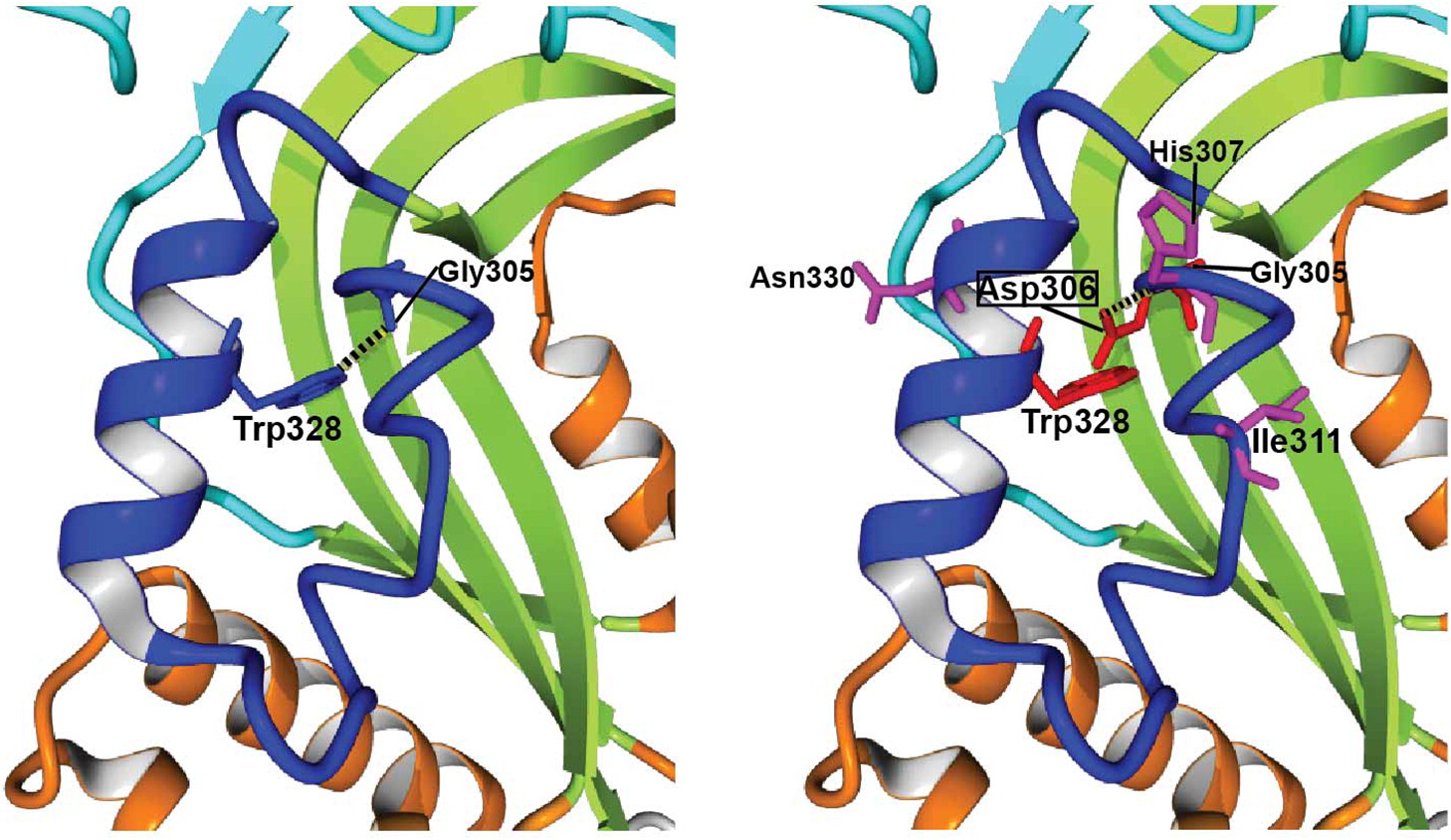
Figure 4, Asp306 creates important steric clashes with Gly305 and Trp328, and perturbs the hydrogen bond (H-bond) between wild-type (WT)-Gly305 and WT-Trp328. The latter residue extends from the neighboring helix and is highly conserved both in vertebrate perforins and in other MACPF proteins (
Figure 3), which suggests that the H-bond with WT-Gly305 or equivalent glycine plays a critical role in stability. Moreover, p.Gly306Asp originates a new H-bond between Asp306 and His307, which is absent in the WT structure. With these and other mutation-specific alterations not mentioned above, it is very likely that these 3 substitutions, and probably nearly all Gly306 variants, would be deleterious for perforin folding and (or) function.