Interstitial lung disease in infancy: an unusual presentation of CD40 ligand deficiency
Abstract
Introduction: Childhood interstitial lung disease (chILD) syndrome describes findings of respiratory symptoms and diffuse abnormalities on lung imaging. Immunodeficiency and immune dysregulation are increasingly recognized as potential causes of these clinical findings. Patients with CD40 ligand (CD40L) deficiency typically present with sinopulmonary and opportunistic infections secondary to impairments in both humoral and cellular immunity. chILD syndrome has not previously been reported as a presenting feature of this disease.
Objectives: We describe a patient with CD40L deficiency, caused by a novel mutation in CD40LG (c.464 T>A, p.Leu155Gln), who presented in infancy with chILD syndrome and lung biopsy findings of chronic interstitial pneumonitis and patchy pulmonary glycogenosis.
Methods: A left lingula wedge lung biopsy was fixed with formalin and stained for analysis. Whole blood samples from the patient and parents were sent for whole exome sequencing. Flow cytometry to assess CD40L expression on activated CD3+CD8− T cells was performed.
Results: Lung biopsy demonstrated lymphoid aggregates and patchy pulmonary interstitial glycogenosis. Whole exome sequencing revealed maternal inheritance of a novel mutation in CD40LG (c.464 T>A, p.Leu155Gln). Functional studies demonstrated absent CD40L expression on activated CD3+CD8− T cells.
Conclusion: This novel mutation in CD40LG, c.464 T>A, is disease-causing based on the patient’s clinical features and absent CD40L expression. This is the first report of CD40L deficiency presenting as chILD syndrome.
Statement of novelty: The missense mutation observed in our patient (c.464 T>A, p.Leu155Gln), is novel; CD40L deficiency secondary to a different missense variant at the same amino acid position has been described (c.464 T>P, p.Leu155Pro). To our knowledge, this is the first report of CD40L deficiency with an initial clinical picture consistent with interstitial lung disease.
Introduction
Diffuse lung disease is rare in children, and recent classification systems recognize that certain disorders are more prevalent in infants (Vece and Young 2016). chILD (childhood interstitial lung disease) syndrome describes a collection of respiratory symptoms and diffuse abnormalities on imaging (Kurland et al. 2013; Vece and Young 2016). chILD syndrome can be diagnosed if at least 3 of the following 4 criteria are present: (i) respiratory symptoms (cough, dyspnea, exercise intolerance); (ii) respiratory signs (tachypnea, adventitious sounds, retractions); (iii) hypoxemia; and (iv) diffuse abnormalities on a chest radiograph (CXR) or computed tomography (CT) scan (Kurland et al. 2013). These clinical features are associated with a range of underlying pathologies; immunodeficiency and immune dysregulation syndromes are increasingly recognized as causes of chILD syndrome (Vece and Young 2016).
Class switch recombination (CSR) defects are a major group of primary immunodeficiencies. CD40 ligand (CD40L) deficiency, also known as hyper-immunoglobulin M (HIGM) syndrome type 1, is the most common form of CSR syndrome (Korthäuer et al. 1993; Winkelstein et al. 2003; Davies and Thrasher 2010). CD40L deficiency is an X-linked recessive disorder caused by mutations in CD40LG on chromosome Xq26 (Villa et al. 1994); the gene encodes a membrane glycoprotein that is expressed on activated CD4+ T cells. It binds CD40, expressed on B lymphocytes and antigen presenting cells (APC), leading to immunoglobulin class switching, generation of memory B cells, and APC-mediated activation of CD8+ T cells (Noelle et al. 1992; Hollenbaugh et al. 1994).
Affected patients, almost exclusively males due to the X-linked inheritance pattern, present in infancy and early childhood with pulmonary infections due to impaired humoral immunity, and opportunistic infections due to impaired cellular immunity. Autoimmune disorders and malignancies are also common (Davies and Thrasher 2010). Disease phenotype and severity are influenced by the specific gene mutation and degree of protein expression (Buchbinder et al. 2012; Katta et al. 2013). Investigations typically reveal normal to elevated IgM, and low or undetectable IgG, IgA, and IgE. Responses to protein antigens are impaired, however, IgM isohemagglutinins may be present (Davies and Thrasher 2010). T, B, and NK cell numbers are typically normal, however, specific T cell responses may be impaired. Neutropenia is common.
Chronic pulmonary changes, including interstitial lung disease, have been described in patients with both primary and secondary immunodeficiency. Biopsies of patients with primary immunodeficiency commonly demonstrate follicular bronchiolitis, lymphoid hyperplasia, and lymphoid interstitial pneumonia (Dishop 2011). Such findings on lung biopsy should prompt more detailed immunologic investigations. Diffuse pulmonary interstitial glycogenosis (PIG) has been described as a primary interstitial lung disease in infants (Canakis et al. 2002), but patchy PIG is increasingly recognized in association with lung growth abnormalities and cardiac disease (Deterding 2010). It has not previously been described in association with immunodeficiency. Here, we describe a patient with CD40L deficiency, caused by a novel mutation, who presented in infancy with chILD syndrome and lung biopsy findings of chronic interstitial pneumonitis with patchy PIG and mild lymphoid hyperplasia.
Functional and clinical presentation
The patient was the first and only child to healthy, non-consanguineous, Caucasian parents. He was born at 41 weeks via caesarean section for cephalopelvic disproportion. Birth weight was 4260 g. He was formula fed with good growth. Immunizations, including rotavirus, were given without incident. There was no family history of primary immunodeficiency, recurrent infections, early childhood death, autoimmunity, or malignancy.
He presented at 3 months of age following 2 weeks of poor oral intake, weight loss and tachypnea. His weight was 6.15 kg (25–50 percentile). He was tachypneic with mild hypoxia responsive to supplemental oxygen (0.5 L/min). Chest X-ray demonstrated hyperinflation, prominent perihilar markings, and bilateral subsegmental consolidation. Thymic tissue was visible and cardiac silhouette was normal. He was managed with supplemental oxygen, nasogastric (NG) feeding and 5 days of azithromycin. No infectious causes were identified from nasopharyngeal washing and blood cultures. He was transferred to the tertiary care centre 1 week later for lack of clinical improvement.
Repeat microbiological testing of blood cultures and nasopharyngeal washings was negative. A gastroenterology assessment suggested possible gastroesophageal reflux disease, cow’s milk protein allergy, and oral aversion based on history and physical examination; a contrast study of the upper gastrointestinal tract was normal. Sweat chloride testing, biopsy for primary ciliary dyskinesia, and echocardiogram were all normal. Immunologic workup revealed normal IgM, undetectable IgA, and low IgG, which were initially attributed to the physiologic nadir (Table 1). Anti-tetanus and diphtheria toxoid titres were non-protective after 2-month vaccines. Lymphocyte enumeration was normal.
Table 1:
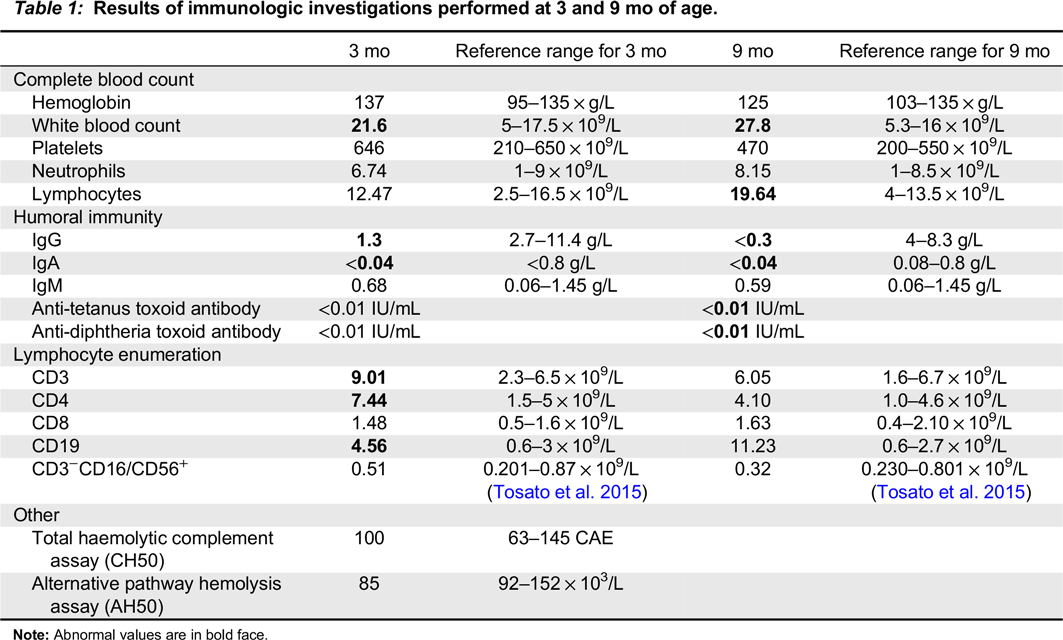
Note: Abnormal values are in bold face.
The patient was treated with amoxicillin, omeprazole and hypoallergenic formula via NG tube. He showed gradual clinical improvement, although tachypnea and mild hypoxia (O2 saturation was 88%–90% on room air) persisted. Persistence of oral aversion necessitated ongoing NG feeds with hypoallergenic formula at discharge.
From 4–6 months of age, growth and stools normalized, but respiratory status remained unchanged. A diagnosis of chILD was considered. The patient underwent a high-resolution chest CT scan performed under sedation, with inspiratory and expiratory acquisitions. This demonstrated typical findings of interstitial lung disease (ground-glass opacities, septal thickening, and air trapping) (Figure 1). There were no pathognomonic features of alveolar proteinosis or Neuroendocrine Cell Hyperplasia of Infancy, the only chILD with recognized specific findings on CT.
Figure 1:
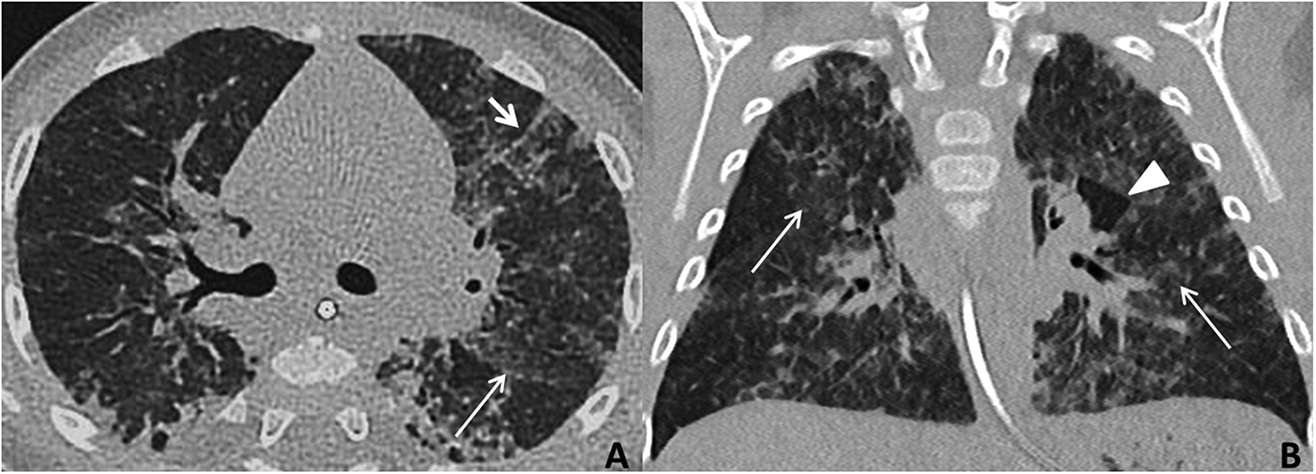
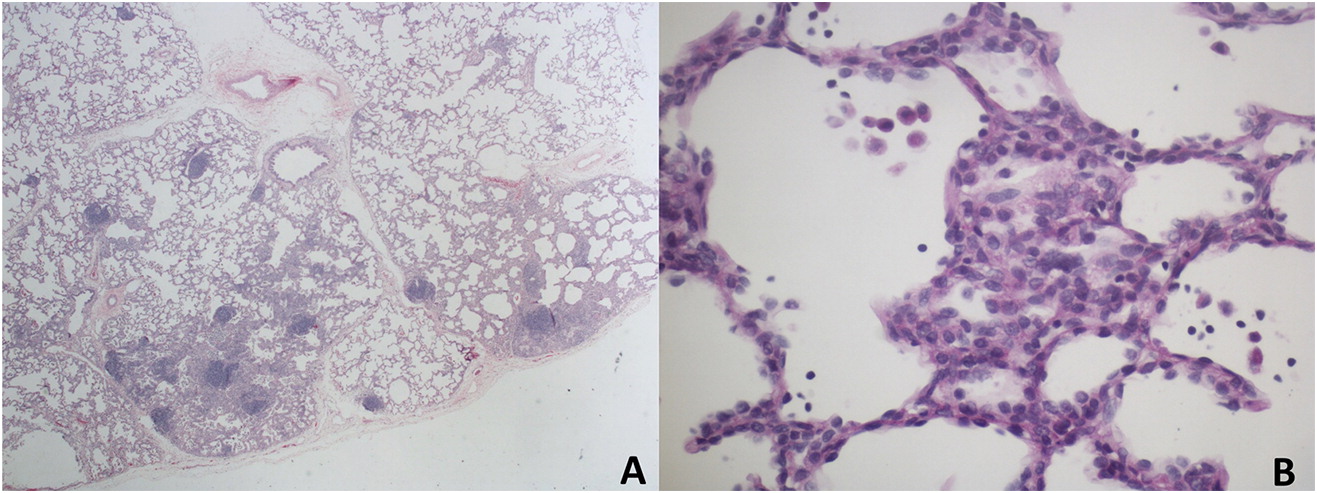
chILD was high on the differential and CT results were not typical of infection, prompting a lung biopsy. A left lingula wedge biopsy of lung was obtained thorascopically, and needle-inflated with formalin for tissue fixation. Formalin-fixed, paraffin-embedded tissue sections were stained with routine hematoxylin and eosin, and special stains for glycogen and pathogenic organisms. Lung pathology showed normal lobular architecture. The alveolar septa in some lobules were abnormally thickened due to an increased number of interstitial mesenchymal cells. A small subset of these cells contained flecks of periodic acid-Schiff (PAS)-positive, diastase-sensitive cytoplasmic glycogen, suggestive of patchy PIG. Scattered lymphoid aggregates consistent with mild lymphoid hyperplasia were also seen (Figure 2). There was evidence of chronic bronchiolar inflammation. There was no viral cytopathic effect, and special stains for Pneumocystis jirovecii (PJP) were negative. The overall impression was that of interstitial pneumonitis of unknown etiology, with a histologic differential diagnosis of lung injury, immunodeficiency, and autoimmune disease.
Figure 2:
At 9 months of age, the patient developed increased tachypnea and hypoxia with worsening of infiltrates on CXR. He was afebrile and otherwise well appearing. Bilateral bronchoalveolar lavage identified PJP, and blood polymerase chain reaction (PCR) identified cytomegalovirus (CMV) viremia (2645 viral copies per mL). Repeat immunologic testing showed undetectable IgG and IgA with normal IgM, and absent vaccine responses to diphtheria and tetanus toxoid, vaccines that had been administered according to schedule at 2, 4, and 6 months. Lymphocyte enumeration remained normal (Table 1). Mitogen stimulation studies revealed normal responses to pokeweed mitogen and Staphylococcus aureus Cowan but reduced T cell response to phytohemagglutinin. Retrospective testing of the newborn blood spot for T-cell receptor excision circles (TRECs) was sent to the Ontario newborn screening program, and returned normal.
Whole blood samples were collected from the patient and both parents and subjected to whole exome sequencing (WES) (GeneDx). This revealed a c.464 T>A mutation in exon 5 of CD40LG, predicted to result in an amino acid substitution, p. Leu155Gln. Sequencing of parent samples confirmed maternal transmission.
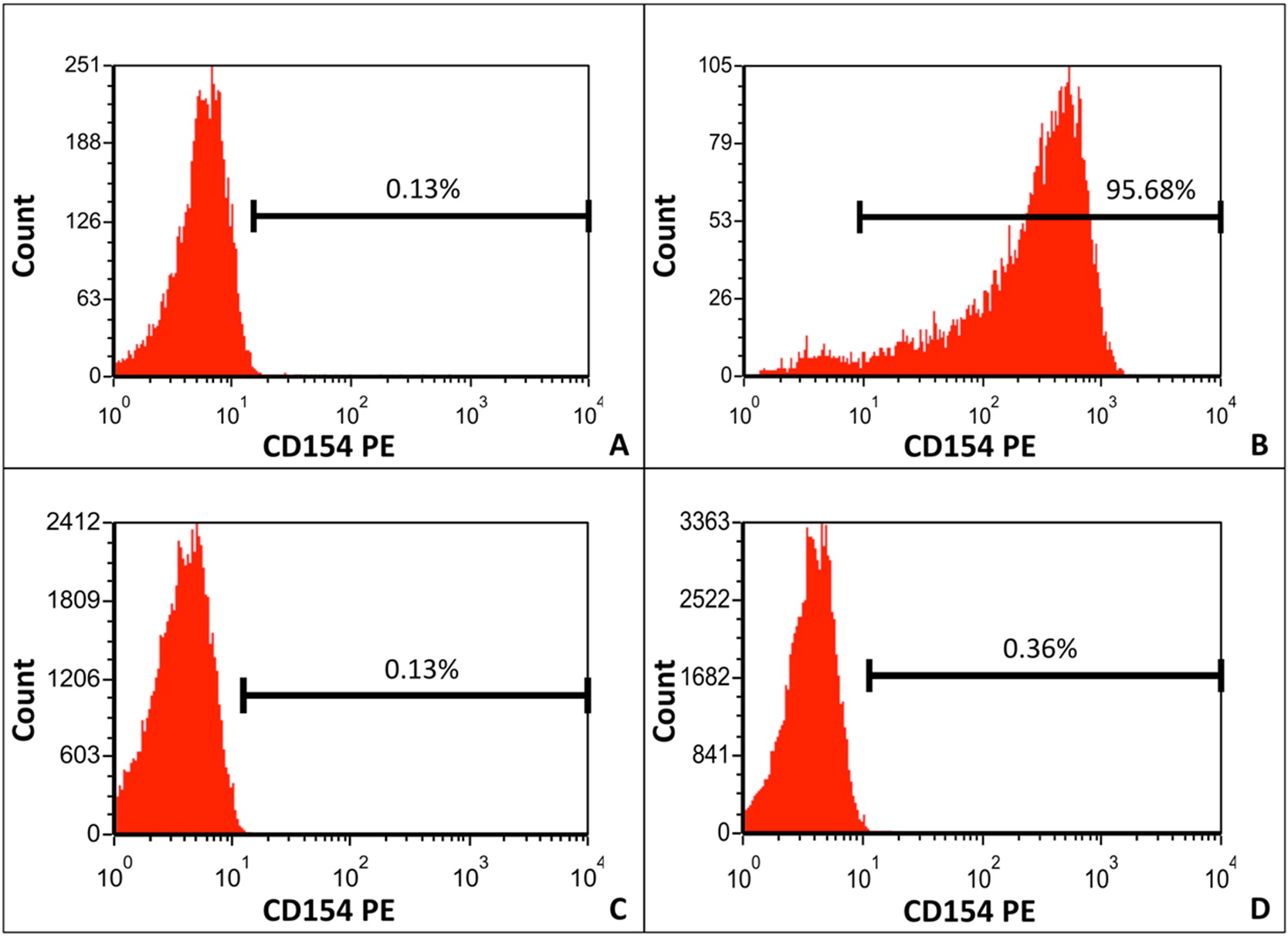
CD40L expression was measured on phorbol myristate acetate (PMA) and ionomycin-stimulated CD3+CD8− T cells using flow cytometry (Calgary Laboratory Services, Alberta, Canada). CD3+CD8− T cells are used because PMA/ionomycin stimulation reduces CD4 antigen density. A healthy procedural control, which was shipped with the patient sample, was run in parallel. CD40L expression on patient PMA and ionomycin-stimulated CD3+CD8− T cells was low (0.36%); expression by the normal control was 95.69% (Figure 3).
Figure 3:
The patient was treated with intravenous trimethoprim-sulfamethoxazole, valgancyclovir, and was started on gammaglobulin replacement therapy. Within a few months of therapy, his respiratory status had improved: he was no longer dyspneic, oxygen saturations had increased, and a CXR was normal in appearance. He is currently doing well following matched unrelated donor bone marrow transplantation.
Discussion
This case describes a patient with CD40L deficiency presenting with chILD syndrome based on tachypnea, hypoxia, failure to thrive, and diffuse abnormalities on chest CT. Identification of the underlying diagnosis was delayed until the patient developed more classic findings of PJP pneumonia and CMV viremia. The identified mutation, c.464 T>A in exon 5 of CD40LG, is extremely likely to be pathogenic based on the patient’s clinical presentation and demonstration of minimal CD40L expression by activated CD3+CD8− T cells. This mutation has not previously been reported, although CD40L deficiency secondary to a different missense variant at the same amino acid position has been described (Lin et al. 1994; Prasad et al. 2005).
The 2013 American Thoracic Society guideline on infant interstitial lung disease suggests evaluation for immunodeficiency in patients presenting with chILD syndrome. Diffuse lung disease in infancy is rare, and the underlying pathology for some of these disorders is distinct from those seen in older children and adults (Vece and Young 2016). chILD syndrome describes a collection of respiratory symptoms including tachypnea, hypoxia, crackles, cough, failure to thrive, and diffuse abnormalities on imaging (Kurland et al. 2013; Vece and Young 2016). The causes of chILD syndrome in infancy are diverse; diagnosis is challenging and relies heavily on pathology (Winkelstein et al. 2003). This patient had pathological findings consistent with patchy PIG. Diffuse PIG has been described in isolation as a primary cause of chILD (Canakis et al. 2002); however, it can also occur along with other conditions such as disorders of lung growth, congenital lung abnormalities and congenital cardiac disease (Deutsch and Young 2010). Interestingly, PIG has only been seen in lung biopsies from young children, suggesting that it may reflect an abnormality in lung development. This case illustrates that PIG can also be seen with the interstitial pneumonitis caused by immunodeficiency and highlights the importance of a careful immunologic workup in children presenting with chILD.
A recent review highlights the evolving role of WES for evaluation of diffuse lung diseases (Vece and Young 2016); recent case reports have identified genetic changes impacting CTLA4 trafficking and STAT3 signaling (Vece and Young 2016). Application of this diagnostic tool earlier in our patient’s course would also have shortened time to diagnosis.
To the best of our knowledge, this is the first published case of CD40L deficiency presenting with chILD syndrome. Our case indicates that this diagnosis should be considered in patients presenting with signs and symptoms of chILD syndrome, and highlights the importance of thorough immunologic workup and follow-up in this patient population. Use of WES in the diagnostic workup of chILD may facilitate earlier diagnosis.
Acknowledgement
No funding was used for the research reported in the manuscript.
REFERENCES
Buchbinder D., Park S., and Nugent D.2012. X-linked hyper IgM syndrome: A novel sequence variant associated with an atypical mild phenotype. J. Pediatr. Hematol. Oncol.34(5):e212–e214.
Canakis A.M., Cutz E., Manson D., and O’Brodovich H.2002. Pulmonary interstitial glycogenosis: A new variant of neonatal interstitial lung disease. Am. J. Respir. Crit. Care Med.165(11):1557–1565.
Davies E.G. and Thrasher A.J.2010. Update on the hyper immunoglobulin M syndromes. Br. J. Haematol.149(2):167–180.
Deterding R.R.2010. Infants and young children with children’s interstitial lung disease. Pediatr. Allergy Immunol. Pulmonol.23(1):25–31.
Deutsch G.H. and Young L.R.2010. Pulmonary interstitial glycogenosis: Words of caution. Pediatr. Radiol.40(9):1471–1475.
Dishop M.K.2011. Paediatric interstitial lung disease: Classification and definitions. Paediatr. Respir. Rev.12(4):230–237.
Hollenbaugh D., Wu L.H., Ochs H.D., Nonoyama S., Grosmaire L.S., Ledbetter J.A., Noelle R.J., Hill H., and Aruffo A.1994. The random inactivation of the X chromosome carrying the defective gene responsible for X-linked hyper IgM syndrome (X-HIM) in female carriers of HIGM1. J. Clin. Invest.94(2):616–622.
Katta A., Hong J., and Knutsen A.P.2013. Hyper immunoglobulin M syndrome in a 15-year-old boy caused by a Gly219Arg missense mutation. Ann. Allergy Asthma Immunol.110(5):391–393.
Korthäuer U., Graf D., Mages H.W., Brière F., Padayachee M., Malcolm S., Ugazio A.G., Notarangelo L.D., Levinsky R.J., and Kroczek R.A.1993. Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature. 361(6412):539–541.
Kurland G., Deterding R.R., Hagood J.S., Young L.R., Brody A.S., Castile R.G., Dell S., Fan L.L., Hamvas A., Hilman B.C., Langston C., Nogee L.M., and Redding G.J.2013. An official American Thoracic Society clinical practice guideline: Classification, evaluation, and management of childhood interstitial lung disease in infancy. Am. J. Respir. Crit. Care Med.188(3):376–394.
Lin Q., Rohrer J., Allen R.C., Larché M., et al.1994. A single strand conformation polymorphism study of CD40 ligand. J. Clin. Invest. (24).
Noelle R.J., Roy M., Shepherd D.M., Stamenkovic I., Ledbetter J.A., and Aruffo A.1992. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. USA. 89:6550–6554.
Prasad M.L., Velickovic M., Weston S.A., and Benson E.M.2005. Mutational screening of the CD40 ligand (CD40L) gene in patients with X linked hyper-IgM syndrome (XHIM) and determination of carrier status in female relatives. J. Clin. Pathol.58(1):90–92.
Tosato F., Bucciol G., Pantano G., Putti M.C., Sanzari M.C., Basso G., and Plebani M.2015. Lymphocytes subsets reference values in childhood. Cytometry A. 87(1):81–85.
Vece T.J. and Young L.R.2016. Update on diffuse lung disease in children. Chest. 149(3):836–845.
Villa A., Notarangelo L.D., Di Santo J.P., Macchi P.P., Strina D., Frattini A., Lucchini F., Patrosso C.M., Giliani S., Mantuano E., et al.1994. Organization of the human CD40L gene: Implications for molecular defects in X chromosome-linked hyper-IgM syndrome and prenatal diagnosis. Proc. Natl. Acad. Sci. USA. 91(6):2110–2114.
Winkelstein J.A., Marino M.C., Ochs H., Fuleihan R., Scholl P.R., Geha R., Stiehm E.R., and Conley M.E.2003. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine (Baltimore). 82(6):373–384.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 2 • June 2017
Pages: 63 - 69
History
Received: 9 March 2017
Accepted: 9 May 2017
Accepted manuscript online: 21 June 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Victoria E.Cook, Connie L.Yang, Anna F.Lee, AlexiaDabadie, JoanneLuider, Kyla J.Hildebrand, AnneJunker, and Stuart E.Turvey. 2017. Interstitial lung disease in infancy: an unusual presentation of CD40 ligand deficiency. LymphoSign Journal.
4(2): 63-69. https://doi.org/10.14785/lymphosign-2017-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
