Hematopoietic stem cell transplantations for primary immune deficiencies associated with NFκB mutations: a review of the literature
Abstract
Introduction

X-linked ectodermal dysplasia with immunodeficiency
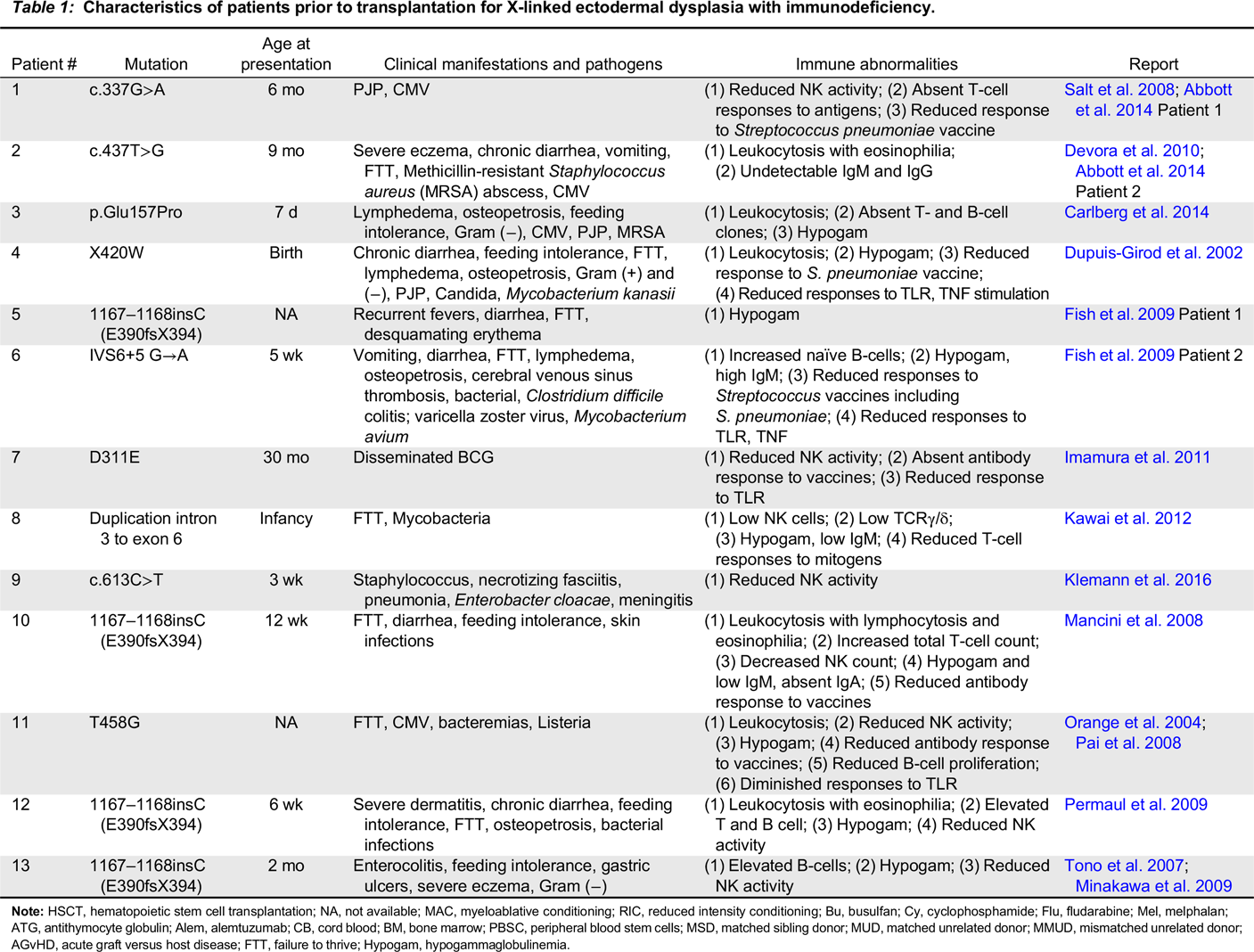
Note: HSCT, hematopoietic stem cell transplantation; NA, not available; MAC, myeloablative conditioning; RIC, reduced intensity conditioning; Bu, busulfan; Cy, cyclophosphamide; Flu, fludarabine; Mel, melphalan; ATG, antithymocyte globulin; Alem, alemtuzumab; CB, cord blood; BM, bone marrow; PBSC, peripheral blood stem cells; MSD, matched sibling donor; MUD, matched unrelated donor; MMUD, mismatched unrelated donor; AGvHD, acute graft versus host disease; FTT, failure to thrive; Hypogam, hypogammaglobulinemia.
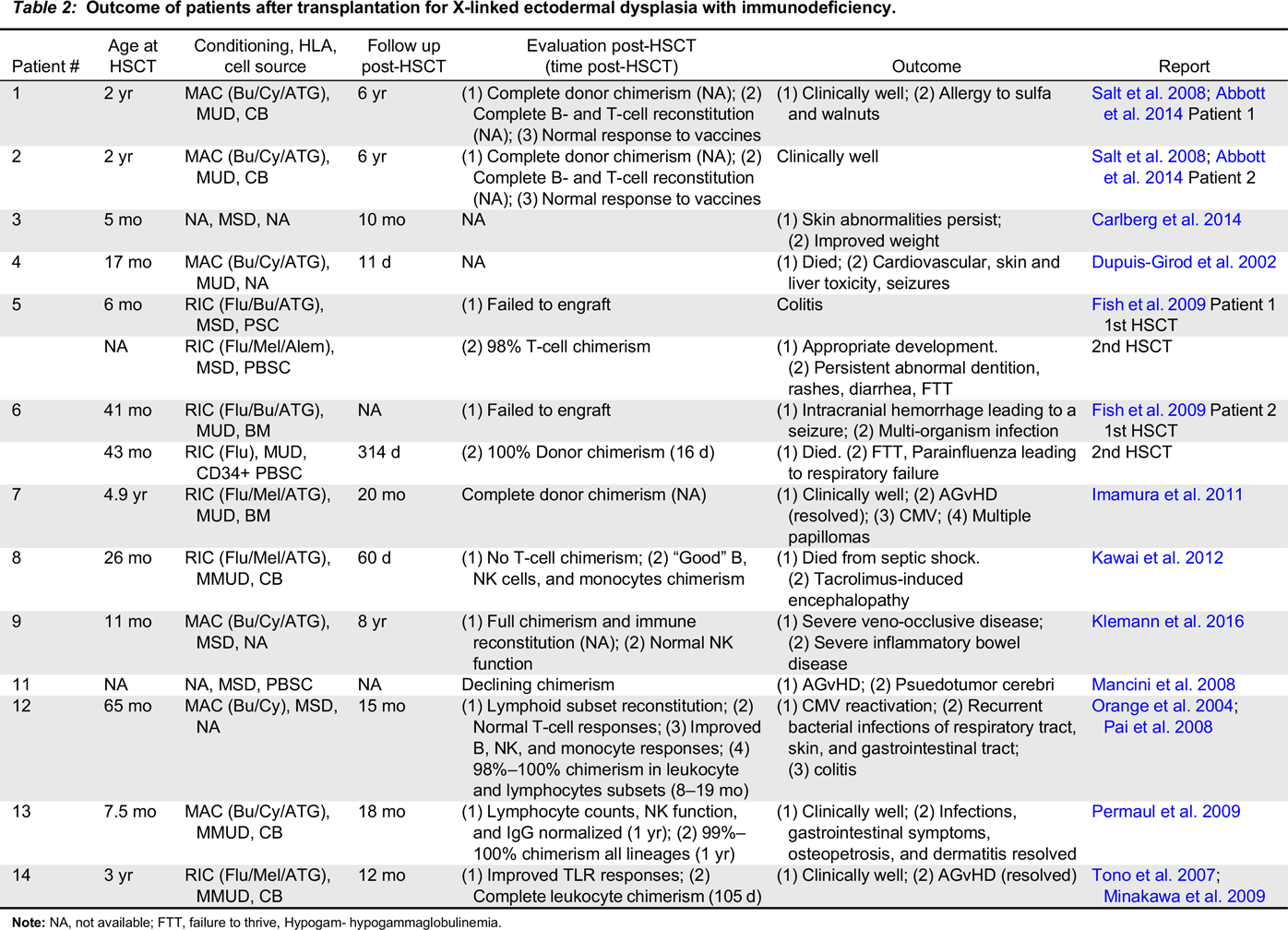
Note: NA, not available; FTT, failure to thrive, Hypogam- hypogammaglobulinemia.
Autosomal dominant ectodermal dysplasia with immunodeficiency
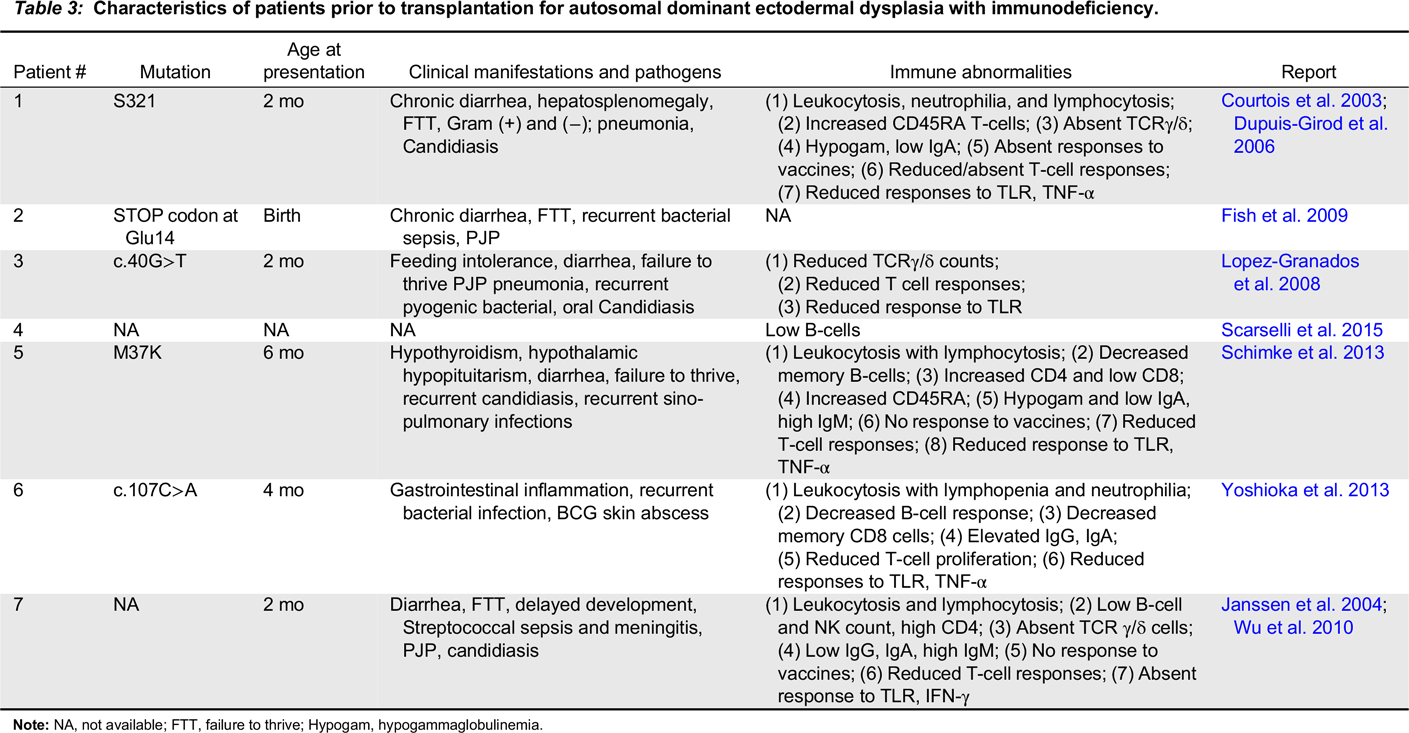
Note: NA, not available; FTT, failure to thrive; Hypogam, hypogammaglobulinemia.
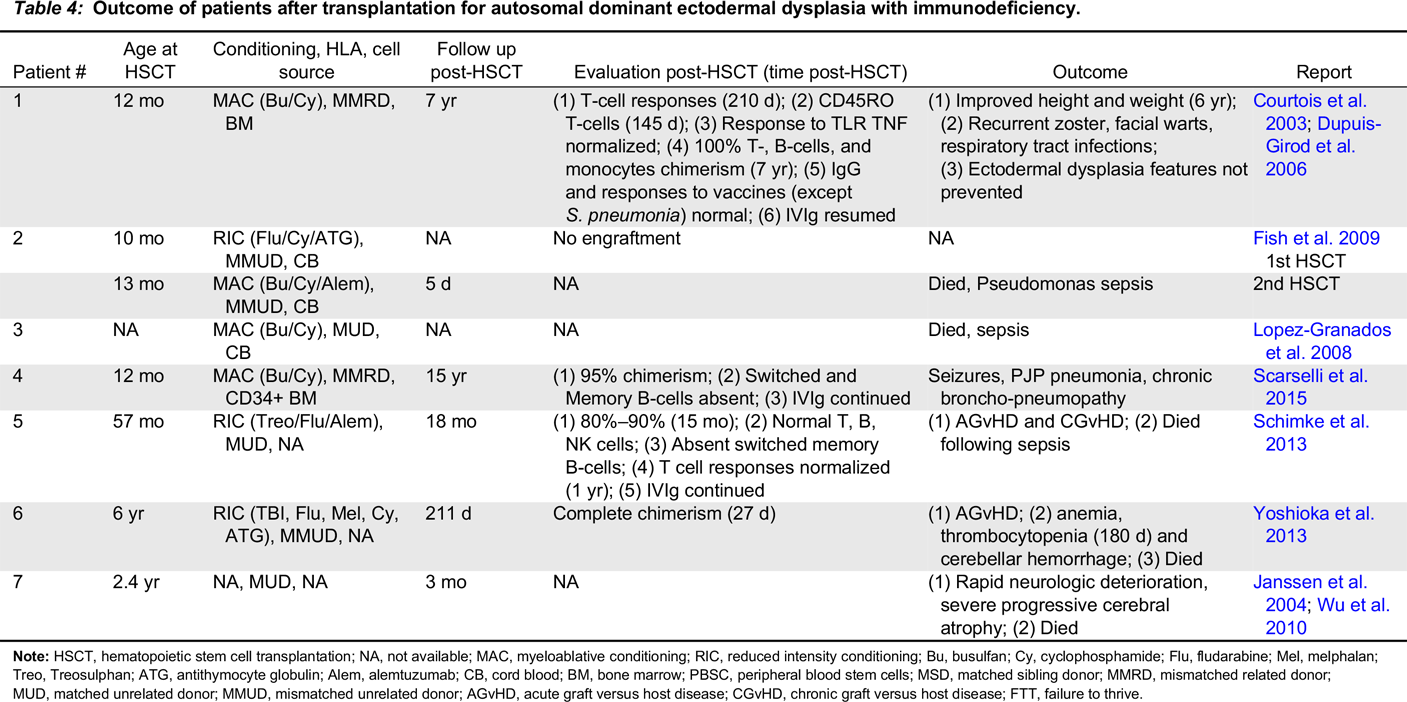
Note: HSCT, hematopoietic stem cell transplantation; NA, not available; MAC, myeloablative conditioning; RIC, reduced intensity conditioning; Bu, busulfan; Cy, cyclophosphamide; Flu, fludarabine; Mel, melphalan; Treo, Treosulphan; ATG, antithymocyte globulin; Alem, alemtuzumab; CB, cord blood; BM, bone marrow; PBSC, peripheral blood stem cells; MSD, matched sibling donor; MMRD, mismatched related donor; MUD, matched unrelated donor; MMUD, mismatched unrelated donor; AGvHD, acute graft versus host disease; CGvHD, chronic graft versus host disease; FTT, failure to thrive.
IKK2 immunodeficiency

Note: NA, not available; FTT, failure to thrive; Hypogam, hypogammaglobulinemia.
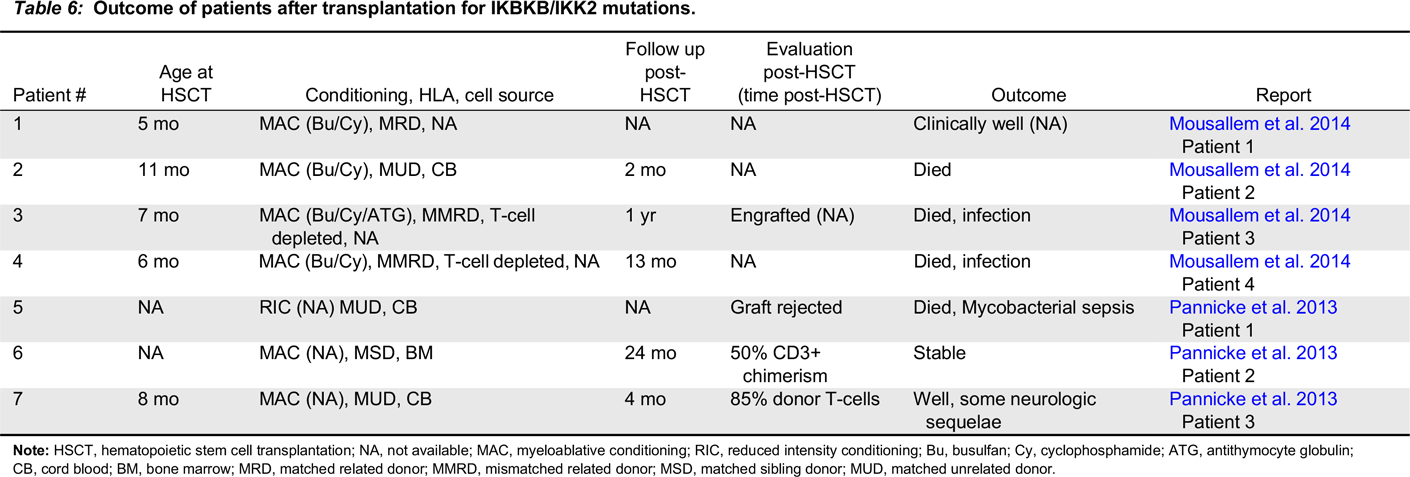
Note: HSCT, hematopoietic stem cell transplantation; NA, not available; MAC, myeloablative conditioning; RIC, reduced intensity conditioning; Bu, busulfan; Cy, cyclophosphamide; ATG, antithymocyte globulin; CB, cord blood; BM, bone marrow; MRD, matched related donor; MMRD, mismatched related donor; MSD, matched sibling donor; MUD, matched unrelated donor.
CARD11-BCL10-MALT1 complex immunodeficiency
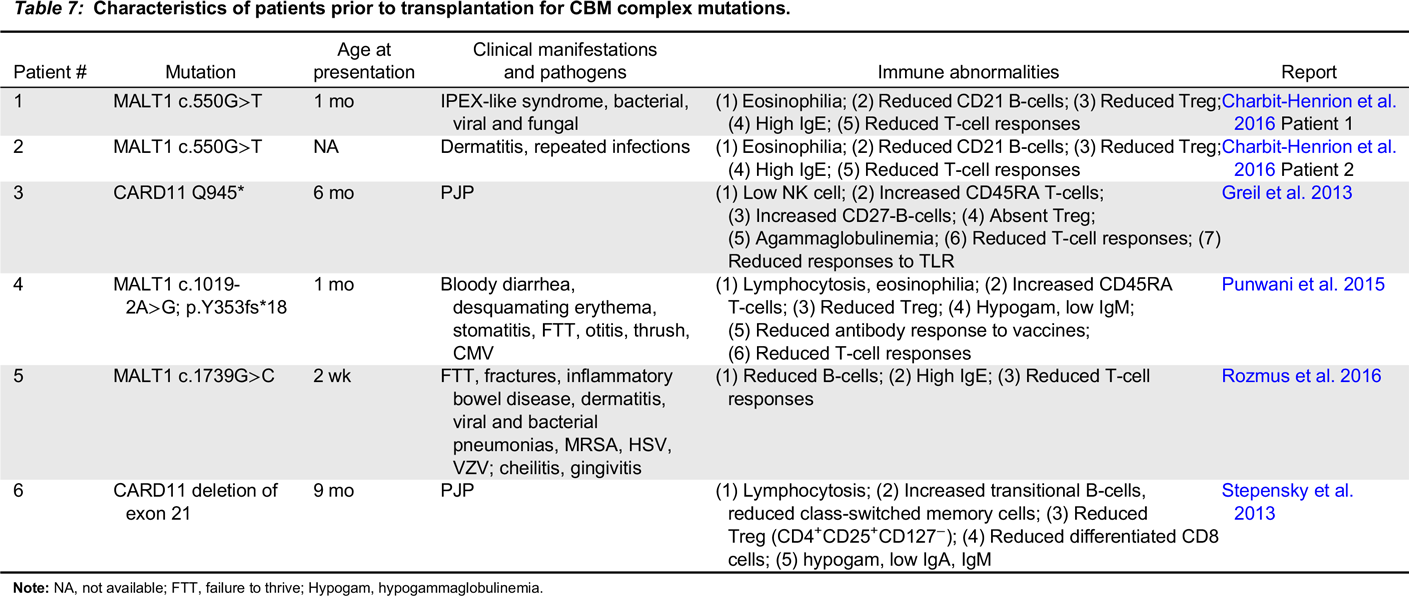
Note: NA, not available; FTT, failure to thrive; Hypogam, hypogammaglobulinemia.
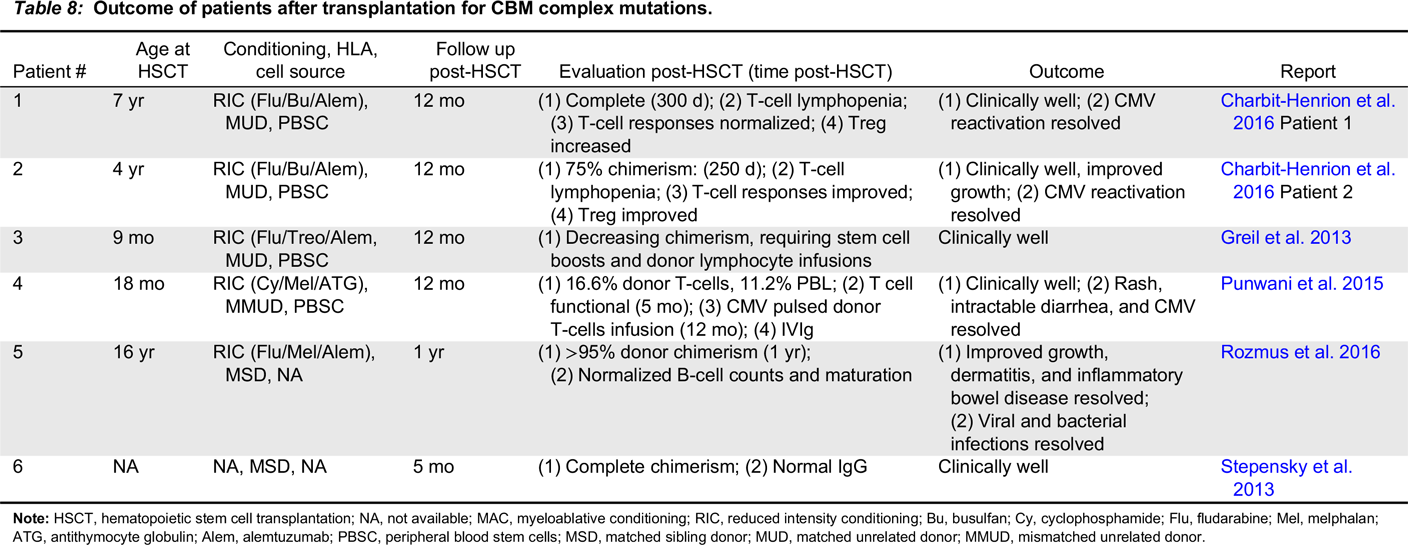
Note: HSCT, hematopoietic stem cell transplantation; NA, not available; MAC, myeloablative conditioning; RIC, reduced intensity conditioning; Bu, busulfan; Cy, cyclophosphamide; Flu, fludarabine; Mel, melphalan; ATG, antithymocyte globulin; Alem, alemtuzumab; PBSC, peripheral blood stem cells; MSD, matched sibling donor; MUD, matched unrelated donor; MMUD, mismatched unrelated donor.
NIK immunodeficiency
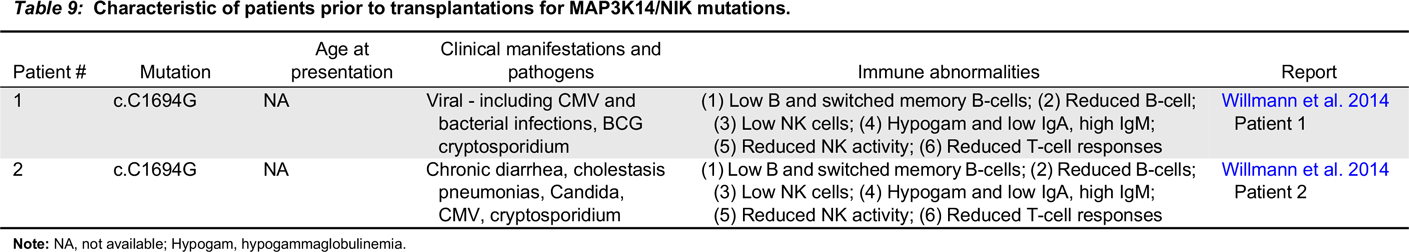
Note: NA, not available; Hypogam, hypogammaglobulinemia.

Note: HSCT, hematopoietic stem cell transplantation; NA, not available; RIC, reduced intensity conditioning; MRD, matched related donor.
Discussion
Conclusion
REFERENCES
Information & Authors
Information
Published In

History
Copyright
Authors
Funding Information
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
