Abnormal apoptosis causes colitis and enteritis in patients with Hoyeraal–Hreidarsson syndrome due to DKC1 mutations
Abstract
Background: Hoyeraal–Hreidarsson syndrome (HHS), the severe clinical variant of X-linked dyskeratosis congenita, is caused by germline mutations in telomere associated genes. HHS usually manifests within the first years of life and is characterized by progressive bone marrow failure, immunodeficiency, neurological features including microcephaly and developmental delay, as well as intrauterine growth retardation. The typical mucocutaneous manifestations are nail dysplasia, lacy skin pigmentation, and oral leukoplakia. Importantly, gastrointestinal involvement is reported in most patients with HHS, and may be the presenting feature. Given the spectrum of gastrointestinal diseases with a similar presentation, recognizing the unique gastrointestinal histopathology of HHS may facilitate earlier diagnosis and treatment.
Methods: This case series highlights the gastrointestinal pathology findings of 2 patients with HHS caused by DKC1 gene mutations.
Results: Gastrointestinal biopsies reveal loss of mucosal glands, regenerative glandular alterations and increased colonic epithelial cell apoptosis. Immunostaining of biopsies for cleaved caspase 3, a marker of cellular apoptosis, demonstrates abnormal nonapical and random locations of enterocyte exit which was further exacerbated by enteritis.
Conclusion: Gastrointestinal involvement is usually the presenting feature of patients with HHS. This case series highlights the important role on gastrointestinal histopathology in facilitating a diagnosis of HHS.
Statement of novelty: Detailed gastrointestinal biopsy images associated with HHS involving DKC1 mutations.
Introduction
Hoyeraal–Hreidarsson syndrome (HHS) is the severe clinical variant of X-linked dyskeratosis congenita (Hoyeraal et al. 1970; Hreidarsson et al. 1988) and is typically caused by germline mutations in telomere associated genes (Glousker et al. 2015). Mutations in the telomerase factors TERT, TPP1, Dyskerin, TIN2, and RTEL1 were reported in patients with HHS, linking impaired telomere maintenance with the disease (Kirwan and Dokal 2009; Zhong et al. 2011; Touzot et al. 2012). In the majority of patients, HHS is caused by mutations in the X-linked dyskeratosis congenita gene, DKC1, encoding Dyskerin (Knight et al. 1999; Borggraefe et al. 2009).
HHS usually manifests within the first years of life and is characterized by progressive bone marrow failure, immunodeficiency, neurological features including microcephaly and developmental delay, as well as intrauterine growth retardation (IUGR) (Revy et al. 2000). The typical mucocutaneous manifestations of HHS are nail dysplasia, lacy skin pigmentation, and oral leucoplakia, which may be present at diagnosis or develop over time (Borggraefe et al. 2009; Glousker et al. 2015).
Gastrointestinal involvement is common in patients with HHS and may be the presenting feature. It poses a significant challenge to supportive treatments (Sznajer et al. 2003). Such manifestations are diverse and include feeding difficulties, enteropathy, colitis, and esophageal dysmotility and strictures (Ohga et al. 1997; Sznajer et al. 2003; Borggraefe et al. 2009; Touzot et al. 2012). In a report summarizing 25 cases of HHS, diarrhea was the initial presentation in almost 30% patients (Borggraefe et al. 2009).
Duodenal and colonic biopsies from patients with HHS demonstrate an alteration in glandular architecture, with a focal rarefaction of the gland leading to glandular atrophy (Sznajer et al. 2003; Borggraefe et al. 2009). These changes are thought to be caused by a decrease in the proliferative abilities of the digestive epithelial cells due to telomere abnormalities (Sznajer et al. 2003; Borggraefe et al. 2009).
Given the spectrum of gastrointestinal diseases with a similar presentation, the lack of awareness of the gastrointestinal histopathology findings in patients with HHS may delay early diagnosis and treatment.
Here we report on the gastrointestinal pathology of 2 patients with HHS who presented with colitis. In both cases, the diagnosis was delayed until more features of the disease became apparent. While a loss of mucosal glands and increased apoptosis was noted, we also detected abnormal non-apical and random exit of enterocytes, which was exacerbated by enteritis.
Case presentation
Patient 1
Patient 1 is reviewed in detail in Marcus (2016).
Patient 2
Patient 2 was born at 33 weeks of gestation by cesarean section due to IUGR and oligohydramnios, with a birth weight of 1.3 kg. He is the first child of non-consanguineous Caucasian parents, with no history of previous miscarriages. The mother was noted to have hypothyroidism, eczema, and vaginal candidiasis, and the father had hyperthyroidism and pityriasis rosea. A family history of early death in childhood due to pneumonia, in 2 siblings of the maternal grandfather, was reported.
After birth, the patient had a prolonged 5-week course of hospitalization for feeding difficulties. At 14 months of age, he was noted to have a white plaque on the tongue that was not responsive to antiviral, antifungal, or corticosteroid treatment. A biopsy of the lesion demonstrated scar tissue. He also experienced chronic bloody diarrhea and reduced appetite, followed by weight loss and failure to thrive (FTT).
At the age of 3 years, an upper and lower gastrointestinal endoscopy was performed, revealing white esophageal plaques and severe patchy colitis involving the rectum and proximal sigmoid. An esophageal biopsy demonstrated superficial epithelial invasion of Candida hyphae. Systemic antifungal treatment was administered for the esophageal lesions, resulting in partial improvement of his symptoms. His height and weight were both below the third percentile for age and he also had leukoplakia. The patient was developmentally appropriate for age. Immune evaluation demonstrated a normal white blood count of 7.8 (5.0–12.0 × 109/L), mild anemia with hemoglobin levels of 102 (110–10 g/L), and normal platelet levels of 186 (150–400 × 109/L). Neutrophil and lymphocyte counts were normal at 4.25 (1.5–8.5 × 109/L) and 2.34 (2–8 × 109/L), respectively. Monocyte counts were mildly increased at 1.03 (0.05–0.80 × 109/L). Liver enzymes, albumin, total protein, electrolytes, and kidney function were all normal. His IgG level was in the lower range of normal 4.9 (4.5–14.3 g/L), whereas other immunoglobulin levels, including IgE, were normal. Lymphocyte immunophenotyping revealed a borderline low count of CD19 cells at 213 (200–2100 cells/μL) and low count of NK cells at 78 (100–1000 cells/μL). CD4 counts were normal at 1342 (500–2400 cells/μL), CD8 of 826 (300–166 cells/μL), total CD3 of 2371 (900–4500 cells/μL) and normal CD4 to CD8 ratio of 1.6 (0.9–2.9). He had a good immune response to the measles, rubella, and tetanus vaccine and an equivocal antibody titer to mumps and varicella. Isohemagglutinins were poor with anti-A 1:4 and anti-B 1:1. Neutrophil oxidative burst index was normal. In vitro lymphocyte stimulation assay with PHA was robust at 1101 (normal >200). An extended endocrinology evaluation was unremarkable.
Shortly afterwards, the patient developed reticulated hyperpigmented lesions on his neck, axilla, and groin, and nail changes with vertical ridging and lichen planus appearance. Together with the ongoing colitis, FTT, and leukoplakia, these new findings were suggestive of dyskeratosis congenita. Genetic testing confirmed a mutation at the DKC1 gene c.146 C>T predicting an amino acid change of threonine to methionine at position 49 (T49M). This mutation was previously reported to cause HHS (Knight et al. 1999).
Since the diagnosis, the patient continues to demonstrate significant FTT with microcephaly (head circumference is 2–3 standard deviations below mean) and intermittent colitis. Surprisingly, he has no history of recurrent significant infections. Repeat bone marrow biopsies and aspirations demonstrate mild hypocellularity. Ongoing immune evaluations demonstrate normal humoral and cellular function, despite hypogammaglobinemia with low IgG level of 4.8 (5.4–13.6 g/L) and low counts of B and NK cells.
Results
Gastrointestinal biopsies were obtained from both patients on multiple occasions throughout their follow-up.
Patient 1
At 11 months of age, gastrointestinal histopathology of the duodenum was normal (Figure 1A). However, colitis was evident throughout the entire colon with loss of mucosal glands and regenerative glandular alterations. The affected colonic crypt cells showed apoptosis without inflammatory cellular infiltrates (Figures 1B and 1C). At 15 months of age, a marked loss of mucosal glands in the colonic mucosa was noted. Persistent colonic epithelial cell apoptosis with no inflammation and colonic ulceration was also evident (Figure 2).
Figure 1:
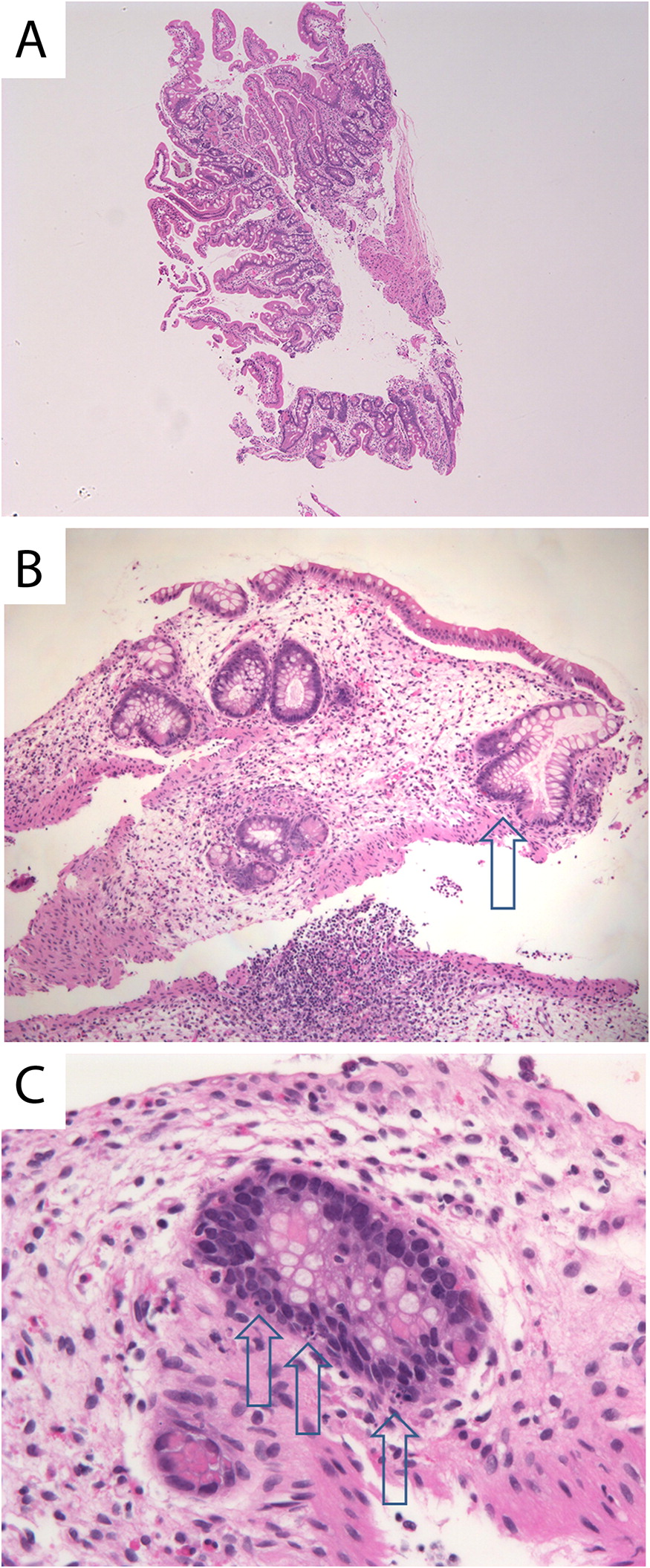
Figure 2:
Patient 2
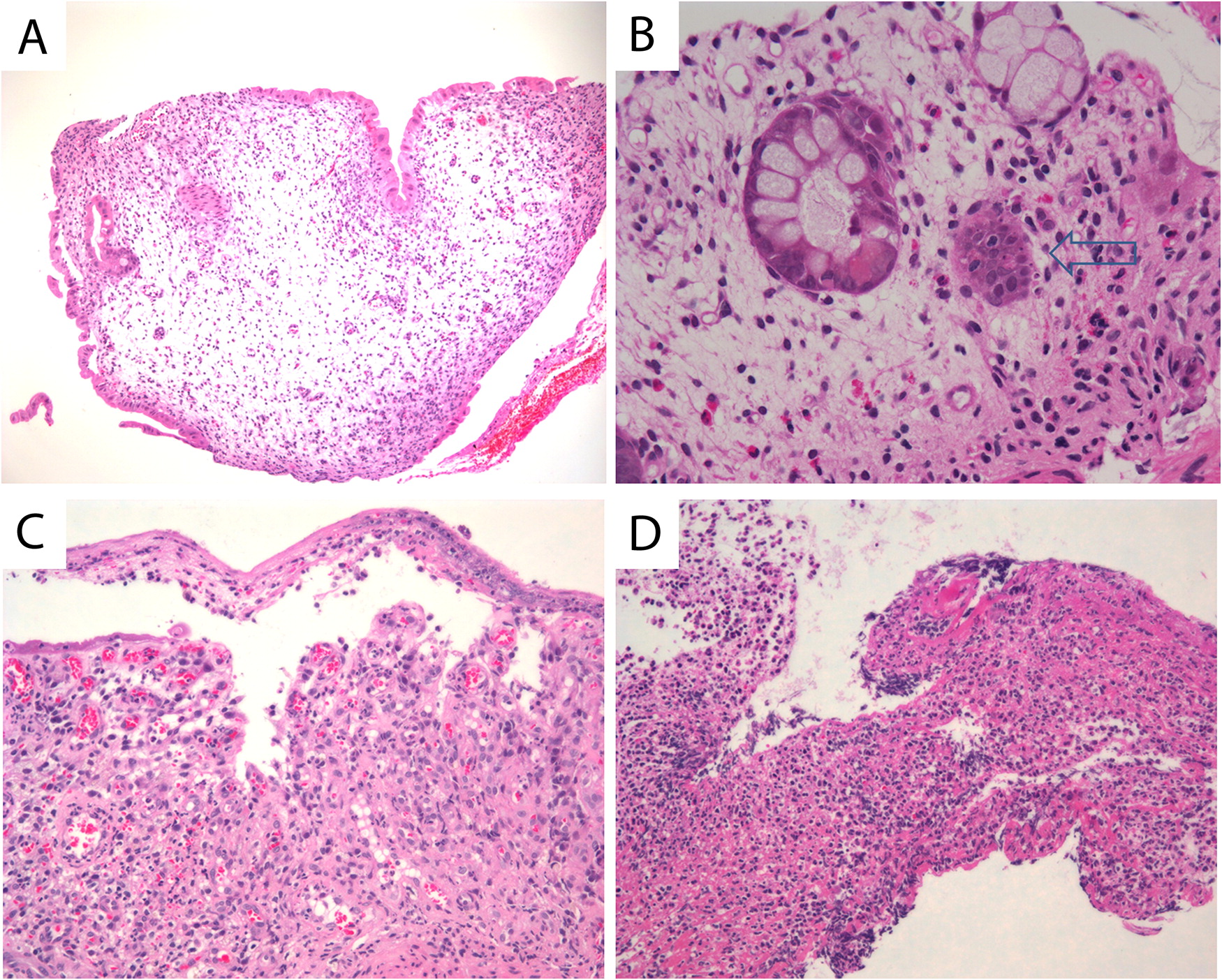
At 5.5 years of age, upper and lower gastrointestinal biopsies demonstrated apoptotic duodenitis without inflammation (Figures 3A and 3B). Sigmoid biopsies revealed focal cryptitis and increased basal crypt apoptosis (Figure 3C).
Figure 3:
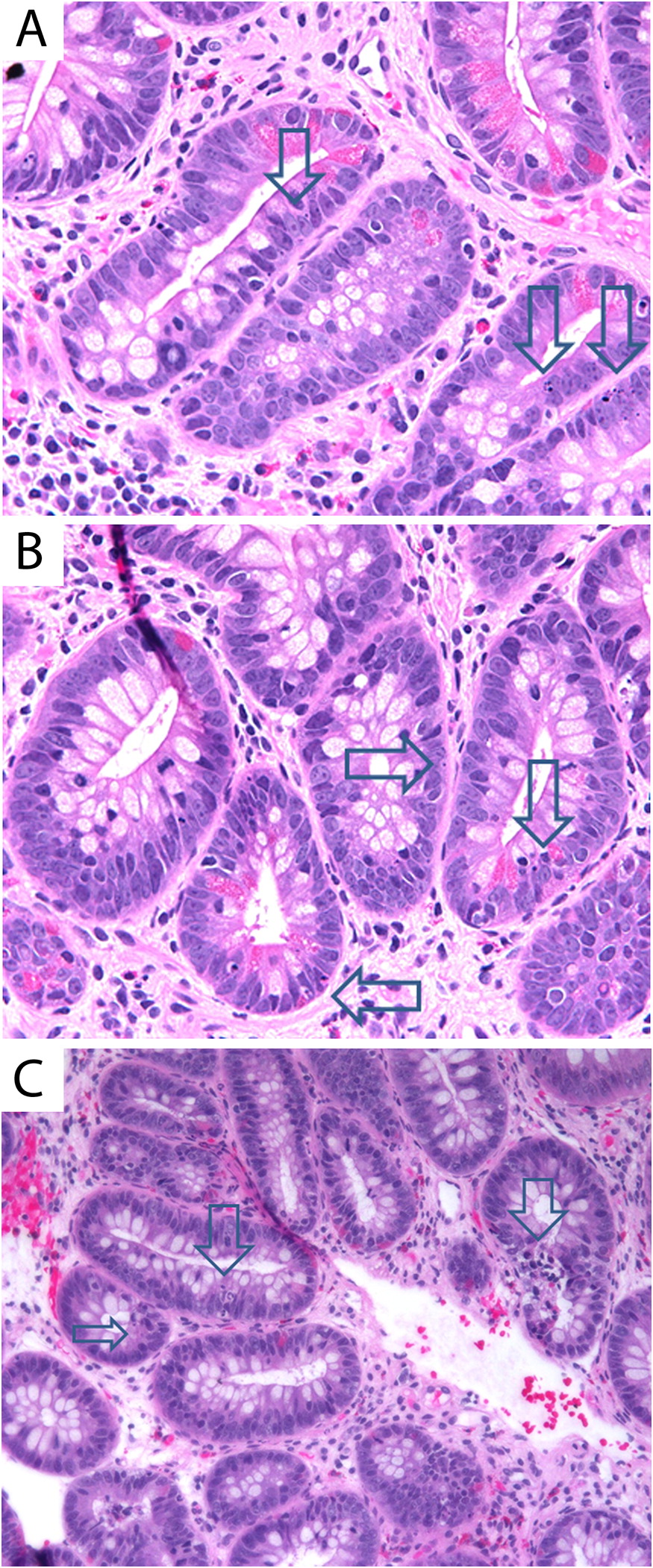
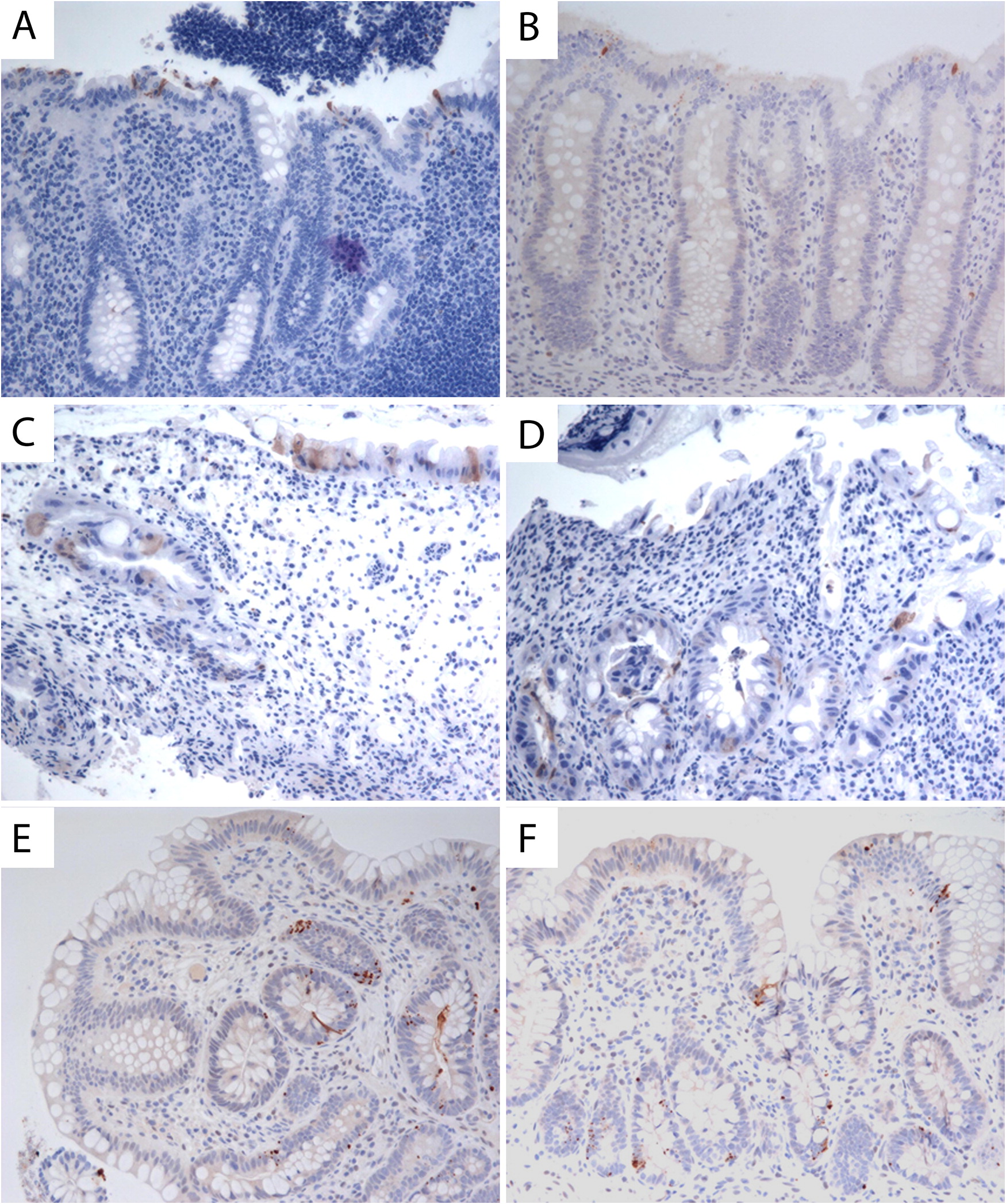
Immunostaining for cleaved caspase 3, a marker of cellular apoptosis, revealed abnormal non-apical locations of enterocyte exit in the gastrointestinal mucosal glands of both patients with HHS. Normal, apical location of enterocyte exit is shown in Figures 4A and 4B. Cleaved caspase 3 immunostaining in biopsies of patient 1 (Figures 4B and 4C) and patient 2 (Figures 4D and 4E) are shown, indicating non-apical and random locations of apoptosis, which was further increased by enteritis in both patients.
Figure 4:
Discussion
HHS is a severe, multisystemic disorder caused by aberrant telomere biology, which mainly affects tissue systems with high cellular turnover, such as the skin, digestive tract, and bone marrow (Sznajer et al. 2003; Glousker et al. 2015).
The patients described in this report have mutations in the DKC1 gene, which encodes the nucleoprotein dyskerin, a cofactor of human telomerase responsible for maintaining telomere length after DNA replication (Dokal 2003; Borggraefe et al. 2009). Telomeres protect chromosome ends, and short dysfunctional telomeres have been shown to induce apoptosis and cells senescence (Armanios and Blackburn 2012).
In this case series, both patients presented with colitis and demonstrate the typical clinical features of HHS, which include IUGR, FTT, and immunodeficiency.
The gastrointestinal pathology findings demonstrate loss of mucosal glands and regenerative glandular alterations throughout the entire colon (Sznajer et al. 2003; Borggraefe et al. 2009). Moreover, abnormal non-apical locations of enterocyte exit and apoptosis were a prominent feature. The lack of inflammation further supports the role of abnormal proliferation in colitis rather than inflammatory processes.
There are several causes of congenital enteritis and FTT, including the loss of normal maintenance of enterocyte adhesion and trafficking, such as apico-basal polarity. For example, patients with tufting enteropathy (Reifen et al. 1994) were reported to have mutations in the EpCAM gene (Sivagnanam et al. 2008), which affects normal intestinal epithelial cell adhesion. Patients with mutations in the TTC7A gene also develop apoptotic colitis (Ngan et al. 2014). This is reportedly due to increased Rho kinase activity causing disruption of polarity, growth, and differentiation of intestinal epithelial cells (Bigorgne et al. 2014). The DKC1 gene defects noted here likely affect telomere function, resulting in dysregulation of the intestinal cell aging process. The histopathological findings of abnormal, non-apical enterocyte exit from the intestinal mucosal glandular crypts, which was further exacerbated by inflammation into non-apical and random enterocyte exit, provides evidence that defects in DKC1 affect the maintenance of the structural intestinal epithelium.
In summary, we reported on 2 patients diagnosed with HHS who presented with early onset colitis. Gastrointestinal manifestations are a significant feature of HHS and in many cases may be the initial presentation. The histopathological findings of increased apoptosis due to abnormal telomere function and maintenance are unique to these patients. Thus, the diagnosis of HHS should be considered in any child presenting with early onset colitis. Awareness of the unique pathological findings may facilitate early diagnosis and management, thereby altering the course of this lethal disease.
REFERENCES
Armanios M. and Blackburn E.H.2012. The telomere syndromes. Nat. Rev. Genet.13(10):693–704.
Bigorgne A.E., Farin H.F., Lemoine R., Mahlaoui N., Lambert N., Gil M., Schulz A., Philippet P., Schlesser P., Abrahamsen T.G., Oymar K., Davies E.G., Ellingsen C.L., Leteurtre E., Moreau-Massart B., Berrebi D., Bole-Feysot C., Nischke P., Brousse N., Fischer A., Clevers H., and de Saint Basile G.2014. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clini. Invest.124(1):328–337.
Borggraefe I., Koletzko S., Arenz T., Fuehrer M., Hoffmann F., Dokal I., Vulliamy T., Weiler V., Griese M., Belohradsky B.H., and Lang T.2009. Severe variant of x-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome) causes significant enterocolitis in early infancy. J. Pediatr. Gastroenterol. Nutr.49(3):359–363.
Dokal I.2003. Inherited aplastic anaemia. Hematol. J.: Off. J. Eur. Haematol. Assoc./EHA. 4(1):3–9.
Glousker G., Touzot F., Revy P., Tzfati Y., and Savage S.A.2015. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br. J. Haematol.170(4):457–471.
Hoyeraal H.M., Lamvik J., and Moe P.J.1970. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr. Scand.59(2):185–191.
Hreidarsson S., Kristjansson K., Johannesson G., and Johannsson J.H.1988. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta. Paediatr. Scand.77(5):773–775.
Kirwan M. and Dokal I.2009. Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys. Acta (BBA) — Mol. Basis Dis.1792(4):371–379.
Knight S.W., Heiss N.S., Vulliamy T.J., Aalfs C.M., McMahon C., Richmond P., Jones A., Hennekam R.C.M., Poustka A., Mason P.J., and Dokal I.1999. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br. J. Haematol.107(2):335–339.
Marcus N.2016. Immunodeficiency and severe gastrointestinal manifestations in a patient with novel DKC1 mutations causing Hoyeraal–Hreidarsson syndrome. LymphoSign J.3(3):xx–xx.
Ngan B., Merico D., Marcus N., Kim V.H.D., Upton J., Bates A., Herbrick J., Nalpathamkalam T., Thiruvahindrapuram B., Cox C., and Roifman C.R.2014. Mutations in tetratricopeptide repeat domain 7A (TTC7A) are associated with combined immunodeficiency with dendriform lung ossification but no intestinal atresia. LymphoSign J.1(1):10–26.
Ohga S., Kai T., Honda K., Nakayama H., Inamitsu T., and Ueda K.1997. What are the essential symptoms in the Hoyeraal-Hreidarsson syndrome?Eur. J. Pediatr.156(1):80–81.
Reifen R.M., Cutz E., Griffiths A.M., Ngan B.Y., and Sherman P.M.1994. Tufting enteropathy: A newly recognized clinicopathological entity associated with refractory diarrhea in infants. J. Pediatr. Gastroenterol. Nutr.18(3):379–385.
Revy P., Busslinger M., Tashiro K., Arenzana F., Pillet P., Fischer A., and Durandy A.2000. A syndrome involving intrauterine growth retardation, microcephaly, cerebellar hypoplasia, B lymphocyte deficiency, and progressive pancytopenia. Pediatrics. 105(3):E39.
Sivagnanam M., Mueller J.L., Lee H., Chen Z., Nelson S.F., Turner D., Zlotkin S.H., Pencharz P.B., Ngan B.Y., Libiger O., Schork N.J., Lavine J.E., Taylor S., Newbury R.O., Kolodner R.D., and Hoffman H.M.2008. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 135(2):429–437.
Sznajer Y., Baumann C., David A., Journel H., Lacombe D., Perel Y., Blouin P., Segura J.F., Cezard J.P., Peuchmaur M., Vulliamy T., Dokal I., and Verloes A.2003. Further delineation of the congenital form of X-linked dyskeratosis congenita (Hoyeraal-Hreidarsson syndrome). Eur. J. Pediatr.162(12):863–867.
Touzot F., Gaillard L., Vasquez N., Le Guen T., Bertrand Y., Bourhis J., Leblanc T., Fischer A., Soulier J., de Villartay J.P., and Revy P.2012. Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J. Allergy Clin. Immunol.129(2):473–482.e3.
Zhong F., Savage S.A., Shkreli M., Giri N., Jessop L., Myers T., Chen R., Alter B.P., and Artandi S.E.2011. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev.25(1):11–16.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 3 • September 2016
Pages: 127 - 133
History
Received: 10 June 2016
Accepted: 11 August 2016
Accepted manuscript online: 11 August 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AdiOvadia and Bo-YeeNgan. 2016. Abnormal apoptosis causes colitis and enteritis in patients with Hoyeraal–Hreidarsson syndrome due to DKC1 mutations. LymphoSign Journal.
3(3): 127-133. https://doi.org/10.14785/lymphosign-2016-0008
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
