Paradoxical hyperhidrosis in a patient with ectodermal dysplasia and immunodeficiency
Abstract
Anhidrotic Ectodermal Dysplasia with Immunodeficiency (EDA-ID) is a pleotropic disorder characterized by dental abnormalities, eccrine sweat dysgenesis, specific facies, fine sparse hair, pale wrinkled skin, and variable immune defects. The condition is caused by hypomorphic mutations (NF) κB Essential modifier protein (NEMO) gene. The clinical phenotype between patients is heterogenous and variable.
Here we report a patient with a known NEMO mutation presenting with clinical features consistent with EDA-ID, except for paradoxical hyperhidrosis despite having a biopsy-proven reduced number of sweat glands.
Statement of novelty: We report a patient with X-linked EDA-ID due to NEMO deficiency who presented with marked diaphoresis despite biopsy-proven reduced sweat glands and ectodermal dysplasia.
Background
Anhidrotic Ectodermal Dysplasia with Immunodeficiency (EDA-ID) is an X-linked pleotropic disorder characterized by dental abnormalities, eccrine sweat dysgenesis, specific facies, fine sparse hair, pale wrinkled skin, and variable immune defects (Orange et al. 2004). It is caused by mutations in the nuclear factor (NF) κB Essential modifier protein (NEMO). NFκB is activated by phosphorylation and degradation of NFκB inhibitors (IκBs) through the enzyme IκB kinase (IKK) (DiDonato et al. 1997; Mercurio et al. 1997; Israël 2000). NEMO is a scaffolding protein for the two catalytic subunits of IKK, IKKα, and IKKβ (Karin and Ben-Neriah 2000; Ghosh and Karin 2002; Orange and Geha 2003).
Various mutations in the gene encoding NEMO result in different clinical phenotypes based on the type, location, and extent of the mutation (Orange et al. 2004). Heterozygous mutations in the NEMO gene cause X-linked dominant incontinentia pigmenti (Smahi et al. 2000) and Bechet’s (Takada et al. 2010) in female patients. In male patients, loss of function of the NEMO gene is lethal in utero (Smahi et al. 2000, 2002; Niehues et al. 2004). Hypomorphic mutations that impair, but do not completely abolish function, lead to the pleotropic disorder EDA-ID, also known as NEMO deficiency (Zonana et al. 2000; Aradhya et al. 2001; Döffinger et al. 2001).
One of the major features of NEMO deficiency is ectodermal dysplasia (ED). The specific role of NFκB activation has been studied in the pathogenesis of ED. ED was initially reported in the literature where cases were linked to the mutations in the ED1/EDA gene, which encodes ectodysplasin, a member of the tumor necrosis factor (TNF) family (Kere et al. 1996; Monreal et al. 1998). Döffinger et al. (2001) conducted experiments utilizing vectors to manipulate the presence of the ectodysplasin gene and examined resulting NFκB activation. They concluded that both NFκB binding and transcriptional activity were induced by ectodysplasin. Ultimately, NEMO-dependent NFκB activation was shown to be an essential step in the ectodsyplasin developmental pathway and, therefore, the 2 conditions share a common pathophysiological mechanism. This finding was further supported as a direct link between NFκB and ectodermal development by Takeda et al. (1999). Mutant mice with a deficiency in the IKK complex had a variety of cutaneous defects similar to ED and also had an inability to activate NFκB in the skin.
Mutations of NEMO are difficult to recognize in males in early infancy, with most patients initially presenting with manifestations of immunodeficiency (Clarke et al. 1987; Döffinger et al. 2001; Orange and Geha 2003). These immune defects may include T-cell dysfunction, NK-cell deficiency, hypogammaglobinemia, and aberrations in innate immunity (Döffinger et al. 2001; Orange et al. 2002; Puel et al. 2004; Hanson et al. 2008). Consequently, patients with NEMO mutations are susceptible to infection by atypical mycobacteria infections, viruses, and pneumocystitis (Hanson et al. 2008).
As patients age, signs and symptoms of ED become more apparent, including the classic triad of sparse hair, conical or missing teeth, and anhidrosis due to lack of or reduced number of sweat glands. Histologically, there is an overall reduction in the size, number, and maturation of eccrine sweat glands, sebaceous glands, and hair follicles in patients with ED (Reed et al. 1970). Anhidrosis can be life-threatening due to significant hyperthermia (Kere et al. 1996; Schneider et al. 2011; Nguyen-Nielsen et al. 2013). It is therefore imperative to inquire about sweating with patients who are suspected to have ED and to include ED in the differential diagnosis of anhidrosis/hypohidrosis (Shelley et al. 1950; Nguyen-Nielsen et al. 2013).
Here, we report a patient with EDA-ID with a confirmed NEMO mutation, who presented with marked diaphoresis despite a biopsy-proven reduction in number of sweat glands. Our case demonstrates that the clinical finding of sweating can be misleading in ruling out EDA-ID as a diagnosis.
Case presentation
A 14-month-old male child presented to the Immunology clinic with recurrent infections and failure to thrive. He was born at 40 weeks to nonconsanguineous parents after an uncomplicated pregnancy. His birth weight was at the 10th percentile. His mother reported he had excessive sweating since birth. She described that his head would be so diaphoretic that the sweat would soak through his pillow or drench the car seat.
During the first year of life, the patient also had recurrent episodes of hyperthermia. He was found to have an E. coli positive urinary tract infection on one occasion and at 13 months had a pneumococcal bacteremia. Evaluation of the remaining episodes of fever failed to yield an identifiable infectious etiology. He had no significant weight loss. The patient did not have any upper respiratory tract symptoms, oral thrush, diarrhea, vomiting, or rashes during these hyperthermic episodes.
His immunizations were up to date. His family history was significant for autoimmune conditions including Behcet’s disease in his mother, diagnosed at 15 years of age, and adult-onset lupus in a paternal aunt. His mother also had abnormal teeth that had been cosmetically corrected.
His examination revealed height and weight under the third percentile and head circumference at the 50th percentile. He had small palpable lymph nodes present in the groin and axilla. The patient was notably diaphoretic on examination. His hair was wet with sweat. Other physical abnormalities noted included 2 conical incisors and carbuncles on his scalp and axillae. The remainder of the physical exam was unremarkable.
Laboratory findings
Laboratory investigations done at the time of consultation revealed an abnormal complete blood count with elevated lymphocytes, low hemoglobin, and elevated platelets. His erythrocyte sedimentation rate was mildly elevated (Table 1). Flow cytometry analysis showed normal numbers of CD20+ B cells and NK cells. There were elevated values of CD3+, CD4+, and CD8+ cells (Figure 1). His serum IgG level was low at 2.1 g/L and IgM was high at 5.05 g/L. IgE was undetectable, and IgA was 0.2 g/L. His specific antibody titers for tetanus toxoid were not protective.
Table 1:
Figure 1:
Genetic defect
Due to his low IgG, high IgM, and conical incisors, NEMO deficiency was suspected. A previously described (van Raam et al. 2009) nonsense mutation was found in the NEMO gene, resulting in a stop codon (p.Q365X) in exon 9 (c1093C>T) in the TNF-like domain, and this was confirmed by a commercial laboratory. He had a normal male karyotype of 46 XY. His mother was confirmed to be a carrier of the same mutation.
Histopathologic findings
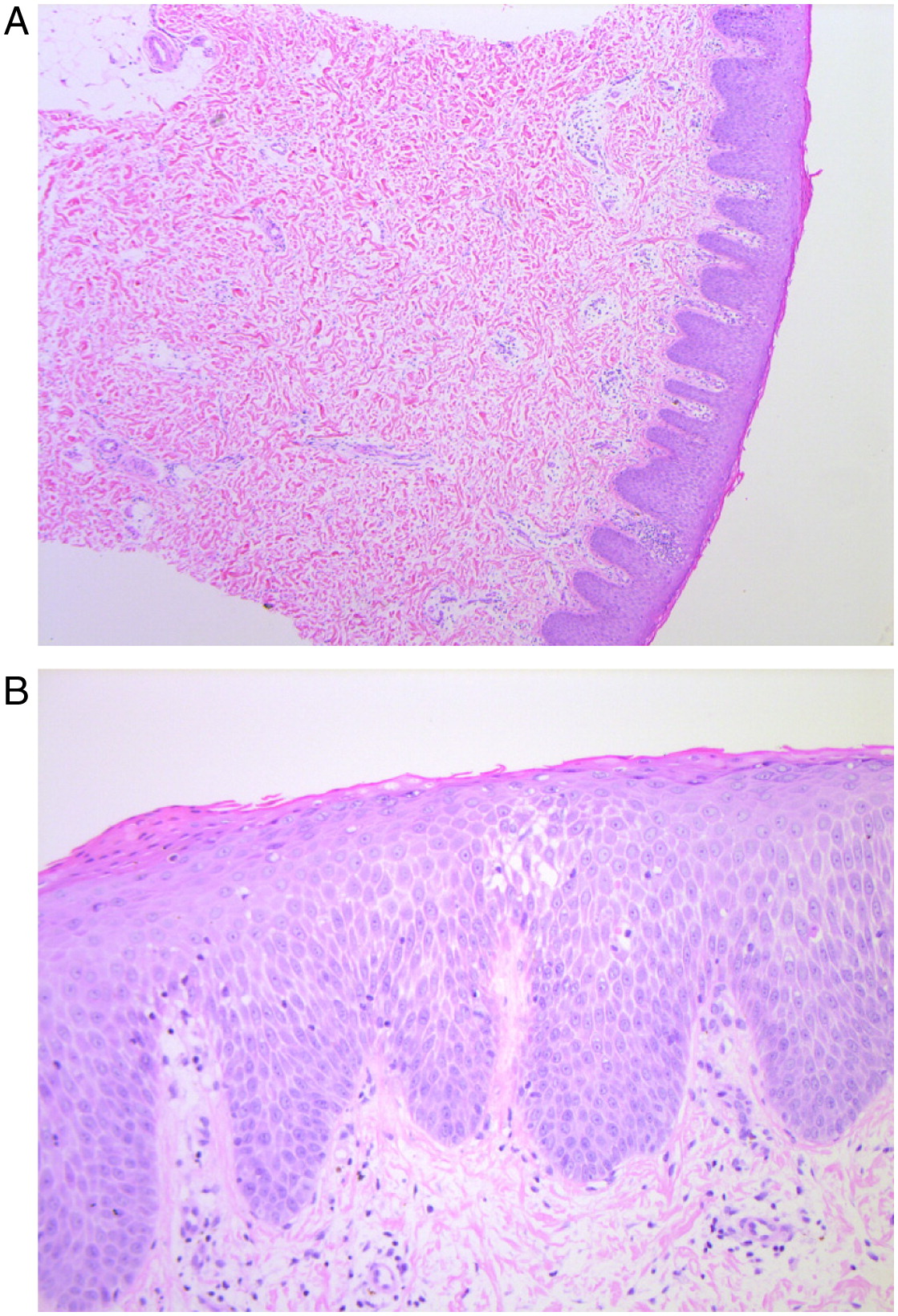
At 2 years of age an 8 mm erythematous nodule with a pale rim was biopsied from his left leg. The punch biopsy demonstrated an absence of hair follicles and eccrine glands (Figure 1a). It also showed psoriasiform hyperplasia and spongiosis with focal parakeratosis, occasional dyskeratotic cells, and mild pigment incontinence (Figure 1b). No eosinophils were seen.
Therapy
The patient was started on monthly IVIG replacement and antimicrobial prophylaxis. The family was counseled about the reduction in sweat glands and measures to reduce the risk of hyperthermia.
Discussion
Patients with ED classically present with anhidrosis or hypohidrosis due to inappropriate ectodermal development. Here, we present a male patient with a NEMO mutation and ED who paradoxically presented with significant hyperhidrosis. There have been previous reports of patients with a NEMO mutation presenting without anhidrotic ectodermal dysplasia (Niehues et al. 2004; Orange et al. 2004); however, hyperhidrosis has not been reported. The previously reported occurrence of the mutation Q365X described a 1-year-old boy with the typical manifestations of EDA-ID including sparse hair, conical incisors, and recurrent bacterial infections although the sweating ability was not specifically discussed (van Raam et al. 2009).
Eccrine sweating is important in human thermoregulation and abnormalities affecting this control can lead to fatal hyperpyrexia (Blüschke et al. 2010). Children with ED are at a substantially increased risk of sudden death due to hyperthermia given their reduced number and function of their sweat glands (Salisbury and Stothers 1981; Clarke et al. 1987). Blüschke et al. (2010) discussed that depending on the climate zone and preventative health care measures, infant mortality for children with ED is between 2% and 20%.
Consistent with patients with EDA-ID studied in Clarke et al. (1987), our patient had a reduced number of sweat glands in the area biopsied, although it is not immediately clear why our patient had hyperhidrosis.
We know that on a molecular basis all boys with EDA-ID must have some functional NEMO protein and therefore some NFκB activity because null mutations are lethal (Puel et al. 2004). The lethality, based on extrapolation from knockout mice lines, is likely secondary to the lack of protection against apoptosis (Rudolph et al. 2000). Different mechanisms have been described to achieve this function and patients with EDA-ID are usually described to have “hypomorphic mutations.” These mutations by definition have reduced activity rather than no activity and it is reasonable to suggest that this residual NFκB activity might lead to inconsistently affected end organ results. Hanson et al. (2008) evaluated the phenotypic diversity found in patients with NEMO. The authors found that certain phenotypes were characteristic of specific NEMO domains, with EDA localizing to mutations in 3 distinct NEMO regions. However, their evaluation of these grouped mutations failed to demonstrate uniform characteristics, which suggests variable penetrance of the NEMO mutation.
Another possibility for variably affected cells in NEMO mutations could be because of some unaffected cells; boys have been described with null NEMO mutations who survived due to Klinefelter’s or somatic mosaicism (Kenwrick et al. 2001). In our case, Klinefelter’s was ruled out by karyotyping. Somatic mosaicism continues to remain a possibility depending on the timing and location of the somatic rearrangement of NEMO, although in our patient it is less likely given the presence of a point mutation rather than the large deletion described in the 3 patients reported by Kenwrick et al. (2001).
We know from other phenotypic features of NEMO that a particular characteristic can be variably affected. For example, Ku et al. (2005) described 2 patients with NEMO who had agenesis of some teeth, whereas other teeth were conical. This observation supports the possibility that our patient may have an absence of sweat glands in one area but may have developed sweat glands elsewhere.
Likely in our case his sweat gland density is variable. We know he had significantly reduced sweat gland numbers in his lower limb biopsy. Perhaps other areas, such as his head, had sweat glands present in adequate numbers. If his sweat gland density was variable then this may explain why he was prone to hyperthermia and diaphoresis. His reduction in sweat glands could have made him prone to overheating, which triggered the localized sweating response which was ineffective to provide adequate cooling.
Our patient was advised how to prevent calamities related to hyperthermia, such as taking refuge in cooler places in hot weather, moistening clothing, avoiding over-exertion, using portable ventilators, or taking cool drinks (Blüschke et al. 2010; Hammersen et al. 2011). These measures are in addition to his immunological treatments. He no longer experiences frequent fevers without a cause.
In conclusion, the identification of a patient with EDA-ID with marked diaphoresis despite biopsy-proven reduced sweat glands presents an additional diagnostic challenge, given that it contrasts with the typical phenotype of hypohidrosis/anhidrosis. The presentation of a child with a NEMO mutation can be subtle, and clinically observed sweating can be paradoxical. The presence of excessive sweating in a patient presenting with immunodeficiency should not be used as evidence to exclude ED.
REFERENCES
Aradhya S., Courtois G., Rajkovic A., Lewis R.A., Levy M., Israël A., and Nelson D.L.2001. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am. J. Hum. Genet.68(3):765–771.
Blüschke G., Nüsken K.D., and Schneider H.2010. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum. Dev.86:397–399.
Clarke A., Phillips I., Brown R., and Harper P.1987. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch. Dis. Childhood.67:989–996.
DiDonato J.A., Hayakawa M., Rothwarf D.M., Zandi E., and Karin M.1997. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature.388(6642):548–554.
Döffinger R., Smahi A., Bessia C., Geissmann F., Feinberg J., Durandy A., Bodemer C., Kenwrick S., Dupuis-Girod S., Blanche S., and Wood P.2001. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kB signalling. Nat. Genet.27:277–285.
Ghosh S. and Karin M.2002. Missing pieces in the NF-κB puzzle. Cell.109(Suppl):S81–S96.
Hammersen J.E., Neukam V., NüSken K.D., and Schneider H.2011. Systematic evaluation of exertional hyperthermia in children and adolescents with hypohidrotic ectodermal dysplasia: An observational study. Pediatr. Res.70:297–301.
Hanson E.P., Monaco-Shawyer L., Solt L.A., Madge L.A., Banerjee P.P., May M.J., and Orange J.S.2008. Hypomorphic nuclear factor-kB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J. Allergy Clin. Immunol.122(6):1169–1177.
Israël A.2000. The IKK complex: An integrator of all signals that activate NF-κB? Trends Cell Biol.10(4):129–133.
Karin M. and Ben-Neriah Y.2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol.18:621–663.
Kenwrick S., Woffendin H., Jakins T., Shuttleworth S.G., Mayer E., Greenhalgh L., Whittaker J., Rugolotto S., Bardaro T., Esposito T., D'Urso M., Soli F., Turco A., Smahi A., Hamel-Teillac D., Lyonnet S., Bonnefont J.P., Munnich A., Aradhya S., Kashork C.D., Shaffer L.G., Nelson D.L., Levy M., and Lewis R.A.2001. International IP Consortium: Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am. J. Hum. Genet.69(6):1210–1217.
Kere J., Srivastava A.K., Montonen O., Zonana J., Thomas N., Ferguson B., Munoz F., Morgan D., Clarke A., Baybayan P., and Chen E.Y.1996. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Gene.13:409–416.
Ku C.L., Dupuis-Girod S., Dittrich A.M., Bustamante J., Santos O.F., Schulze I., Bertrand Y., Couly G., Bodemer C., Bossuyt X., Picard C., and Casanova J.L.2005. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics.115:e615–e619.
Mercurio F., Zhu H., Murray B.W., Shevchenko A., Bennett B.L., Li J., Young D.B., Barbosa M., Mann M., Manning A., and Rao A.1997. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science.278(5339):860–866.
Monreal A.W., Zonana J., and Ferguson B.1998. Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet.63:380–389.
Nguyen-Nielsen M., Skovbo S., Svaneby D., Pedersen L., and Fryzek J.2013. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur. J. Med. Genet.56(5):236–242.
Niehues T., Reichenbach J., Neubert J., Gudowius S., Puel A., Horneff G., Lainka E., Dirksen U., Schroten H., Döffinger R., Casanova J.L., and Wahn V.2004. Nuclear factor kappaB essential modulator-deficient child with immunodeficiency yet without anhidrotic ectodermal dysplasia. J Allergy Clin. Immunol.114(6):1456–1462.
Orange J. and Geha R.2003. Finding NEMO: Genetic disorders of NF-KB activation. J. Clin. Invest.112:983–985.
Orange J., Jain A., Ballas Z., Schneider L., Geha R., and Bonilla F.2004. The presentation and natural history of immunodeficiency caused by nuclear factor kB essential modulator mutation. J. Allergy Clin. Immunol.113:725–733.
Orange J.S., Brodeur S.R., Jain A., Bonilla F.A., Schneider L.C., Kretschmer R., Nurko S., Rasmussen W.L., Köhler J.R., Gellis S.E., and Ferguson B.M.2002. Deficient natural killer cell cytotoxicity in patients with IKK-c/NEMO mutations. J. Clin. Invest.109:1501–1509.
Priolo M. and Lagana C.2001. Ectodermal dysplasia: A new clinical-genetic classification. J. Med. Genet.38:579–585.
Puel A., Picard C., Ku C.L., Smahi A., and Casanova J.L.2004. Inherited disorders of NF-kappaB-mediated immunity in man. Curr. Opin. Immunol.16:34–41.
Reed W.B., Lopez D.A., and Landing B.1970. Clinical spectrum of anhidrotic ectodermal dysplasia. Arch. Derm.102:134–143.
Rudolph D., Yeh W.C., Wakeham A., Rudolph B., Nallainathan D., Potter J., Elia A.J., and Mak T.W.2000. Severe liver degeneration and lack of NF-kB activation in NEMO/IKKg-deficient mice. Genes Dev.14:854–862.
Salisbury D.M. and Stothers J.K.1981. Hypohidrotic ectodermal dysplasia and sudden infant death. Lancet.1:153–154.
Schneider H., Hammersen J., Preisler-Adams S., Huttner K., Rascher W., and Bohring A.2011. Sweating ability and genotype in individuals with X-linked hypohidrotic ectodermal dysplasia. J. Med. Genet.48(6):426–432.
Shelley W.B., Horvath P.N., and Pillsbury D.M.1950. Anhidrosis: An etiologic interpretation. Medicine. 29(3):195–224.
Smahi A., Courtois G., Rabia S.H., Döffinger R., Bodemer C., Munnich A., Casanova J.L., and Israël A.2002. The NF-jB signalling pathway in human diseases: From incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum. Mol. Genet.11:2371–2375.
Smahi A., Courtois G., Vabres P., Yamaoka S., Heuertz S., Munnich A., Israël A., Heiss N.S., Klauck S.M., Kioschis P., and Wiemann S.2000. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature.405:466–472.
Takada H., Nomura A., Ishimura M., Ichiyama M., Ohga S., and Hara T.2010. NEMO mutation as a cause of familial occurrence of Behçet’s disease in female patients. Clin. Genet.78:575–579.
Takeda K., Takeuchi O., Tsujimura T., Itami S., Adachi O., Kawai T., Sanjo H., Yoshikawa K., Terada N., and Akira S.1999. Limb and skin abnormalities in mice lacking IKKa. Science.284:313–316.
van Raam B.J., van Bruggen R., Tool A.T., Jansen M.H., Warris A., Jolles S., and Kuijpers T.W.2009. Nuclear factor-{kappa}B is not essential for NADPH oxidase activity in neutrophils from anhidrotic ectodermal dysplasia patients. Blood.113(21):5362–5363.
Zonana J., Elder M.E., Schneider L.C., Orlow S.J., Moss C., Golabi M., Shapira S.K., Farndon P.A., Wara D.W., Emmal S.A., and Ferguson B.M.2000. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO)Am. J. Hum. Genet.67:1555–1562.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 2 • June 2016
Pages: 61 - 66
History
Received: 10 February 2016
Accepted: 10 February 2016
Accepted manuscript online: 10 March 2016
Version of record online: 10 March 2016
Copyright
© 2016.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AmiirahAujnarain, CatherineChung, and JuliaUpton. 2016. Paradoxical hyperhidrosis in a patient with ectodermal dysplasia and immunodeficiency. LymphoSign Journal.
3(2): 61-66. https://doi.org/10.14785/lymphosign-2016-0002
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
