Immunodeficiency Canada Primary Immunodeficiency Video Conferences
Various dates, 2014
Multiple sites across Canada
Chronic granulomatous disease with an initial presentation of arthritis and oral ulcers a case report
Mohammed Alsalamah, Mariam Hanna, Chaim Roifman
Division of Immunology, The Hospital for Sick Children, Toronto, ON
Division of Immunology, The Hospital for Sick Children, Toronto, ON
Introduction: Chronic granulomatous disease (CGD) is a rare inherited defect of leukocyte phagocytic function characterized by recurrent infections with catalase producing organisms. Patients with CGD are at risk of inflammatory diseases and frequently lead to a patient's initial presentation. Autoimmune manifestations are reported in up to 6% of patients with CGD (van den Berg et al. 2009). We report on the first case of CGD presenting exclusively with arthritis as the first manifestation of disease.
Case Description: A twelve-year-old Pakistani male of consanguineous parents presented with migratory arthritis, and painless oral ulcerations of six months duration that was minimally responsive to non-steroidal anti-inflammatory treatment. Initial assessment demonstrated elevated inflammatory markers (ESR 62) with an otherwise unremarkable rheumatologic evaluation (ANA 1:40, negative anti-ds-DNA and negative RF). He presented to the emergency department with fevers and arthritis. Repeat serology was significant for a normocytic anemia (Hgb 95), thrombocytopenia (Plt 141), elevated LDH 1603, ferritin 1230 mcg/L, ESR 127, CRP 9.3, hypertiglycerdemia (3.2 mmol/L) and mild transaminitis (ALT 63, AST 87). While the bone marrow did not reveal hemophagocytosis, he had mildly elevated Soluble CD 136 (1,086 ng/mL) and Soluble IL-2-receptor (CD25) (1,698 U/mL). He received a presumptive diagnosis of Macrophage Activation Syndrome given his arthritis history and was treated with oral prednisone with symptom resolution. Within one month, however, the arthritis relapsed and the patient developed a new fever, productive cough and pleuritic chest pain. Chest imaging revealed multiple nodular opacities and enlarged mediastinal lymph nodes and an induced sputum isolated aspergillus fumigates complex prompting screening for primary immunodeficiency. Neutrophil oxidative index was low at 1.26 and 1.48 in keeping with a diagnosis of CGD. Molecular study showed a mutation in NCF1 confirming the diagnosis of Autosomal-recessive CGD.
Discussion: Chronic granulomatous disease can present with an exclusively rheumatologic presentation including arthritis and oral ulceration. CGD should be included in the differential diagnosis of atypical rheumatologic diseases specially those who develop unusual or opportunistic infections. Due to the changing demographics of patients seen in clinical practice, physicians will see unique presentations of rare diseases that been previously only described in literature.
REFERENCE
van den Berg, J.M., van Koppen, E., Ahlin, A., Belohradsky, B.H., Bernatowska, E., Corbeel, L., Español,.T., Fischer, A., Kurenko-Deptuch, M., Mouy, R., Petropoulou, T., Roesler, J., Seger, R., Stasia, M.J., Valerius, N.H., Weening, R.S., Wolach, B., Roos, D., and Kuijpers, T.W. 2009. Chronic granulomatous disease: the European experience. PloS One. 4(4):e5234. PMID: 19381301. doi: 10.1371/journal.pone.0005234.
DOCK8
Alison Haynes
Janeway Children's Hospital and Memorial University of Newfoundland, St. John's, Canada
Janeway Children's Hospital and Memorial University of Newfoundland, St. John's, Canada
Background: Hyper IgE syndrome (HIES) is a primary immunodeficiency with sporadic, autosomal dominant (STAT3 mutation) and autosomal recessive (DOCK8 and TYK2 mutations) inheritance patterns. HIES secondary to DOCK8 mutation is characterized by extensive cutaneous viral and staphylococcal infections, recurrent sinopulmonary infections, severe allergic diseases, increased susceptibility to malignancy, lymphopenia with associated eosinophilia and elevated IgE.
Methods: This case report highlights the clinical presentation and immune investigations of a male patient with a novel DOCK8 mutation.
Results: Our patient presented with cutaneous viral infections including severe molluscum contagiosum and herpes simplex virus plus skin abscesses and acute otitis media. In addition to infections, he developed intermittent diarrhea, nummular eczematous lesions, abnormal fingernails, oral ulcers and Bell's palsy. Immune evaluation revealed lymphopenia, in particular low CD8 cells, low mitogen stimulation response and poor specific antibody production requiring IVIG replacement. Genetic sequencing confirmed a novel mutation in DOCK8.
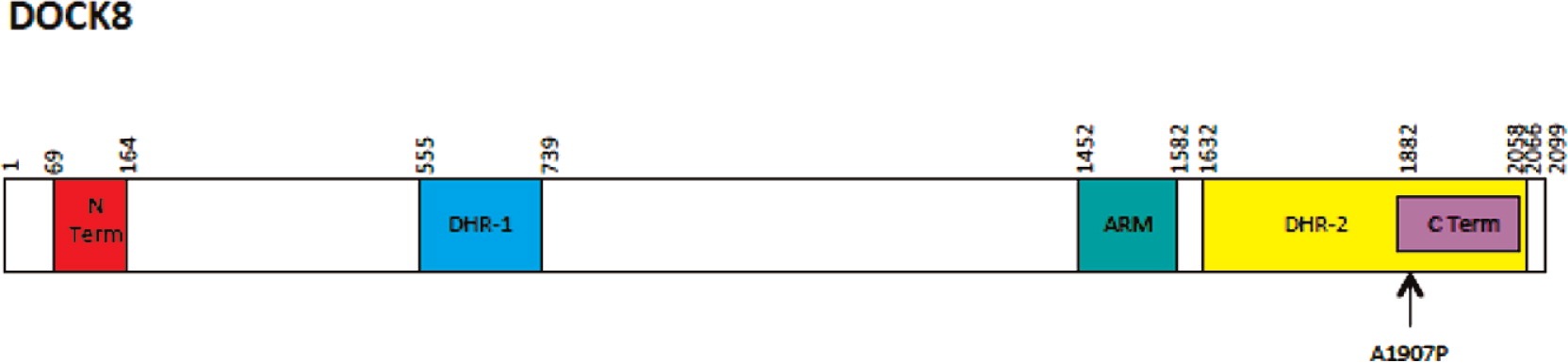
Conclusion: Patients with significant cutaneous viral and bacterial infections, recurrent sinopulmonary infections, severe allergic diseases and lymphopenia with associated elevated IgE should be investigated for DOCK8 mutation. This case report highlights a novel mutation in the DOCK8 gene causing alanine to proline homozygous change A1970 to P1970.
Novelty: Novel mutation in DOCK8
Results:
Male patient from non consanguineous Caucasian family initially referred at age 13 years old for intermittent diarrhea, recurrent molluscum contagiosum, skin abscesses, eczematous skin lesions, and lymphopenia. At age 9 years old, tis patient developed diarrhea occurring every 3–4 months with as many as 10 episodes per day lasting 1 week in duration. The diarrhea episodes were associated with severe abdominal cramping and mucus with absence of blood in stools or associated fever. Infectious history was significant for severe molluscum contagiosum to trunk and extremities beginning as a child and continuing until age 16 years old. Additional viral infections included extensive herpes simplex virus infection limited to the skin at age 23 years old. History of bacterial infections was significant for two skin abscesses located on left arm managed with incision and drainage at age 13 years old and acute otitis media managed with oral antibiotics at age 15 years old. In addition to cutaneous infections, he had widespread nummular eczematous lesions over extremities, trunk and scalp beginning at age 7 years old. These lesions were initially diagnosed as psoriasis and managed with topical corticosteroids with minimal relief. He had abnormal fingernails described as pitting and hypertrophied with associated peeling and crusting around the nailbed. Additional significant findings included intermittent oral ulcers and left sided Bell's palsy. There was no history of allergic diseases.
Family history included healthy non consanguineous mother and father both from the Netherlands. He has an older sister who is healthy. The paternal grandmother had extensive atopic dermatitis. On the maternal side there was a female first cousin with vitilago and another first female cousin with leukemia diagnosed at age 12 years old.
Initial immune investigations were completed at age 13 years old (Tables 1 and 2). There was evidence of poor specific antibody response to tetanus (pre vaccine titer 0.55 IU/mL, post vaccine titer 0.85IU/mL) and pneumococcal (post vaccine titer 31.2 mg/L). Based on the poor specific antibody response, monthly intravenous immunoglobulin was started at age 14 years old. Genetics confirmed novel DOCK8 mutation exon 45 aa1970, c.5908 G>C aa change alanine to proline homozygous change A1970 to P1970 (Figures 1 and 2)
Table 1:
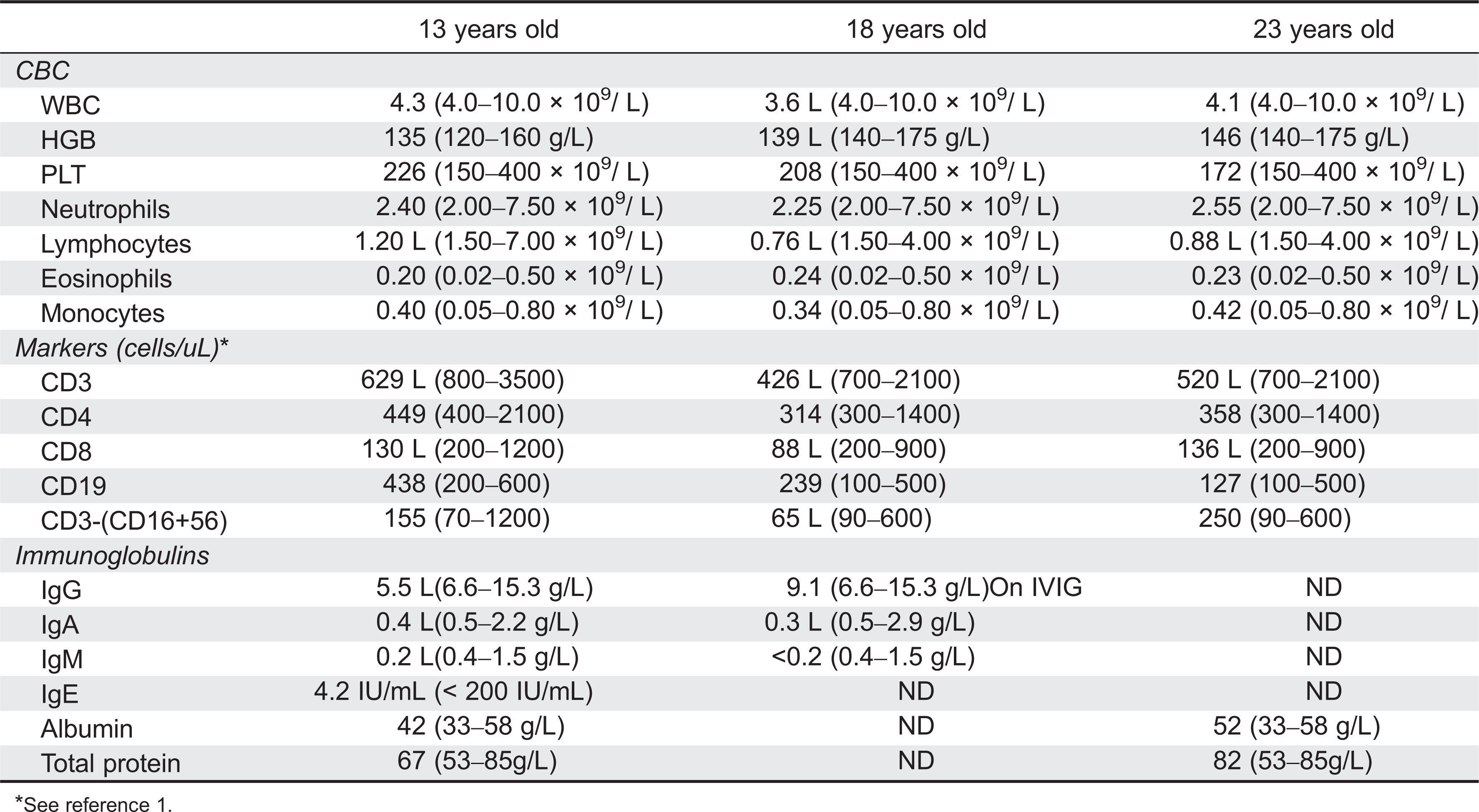
Table 2:
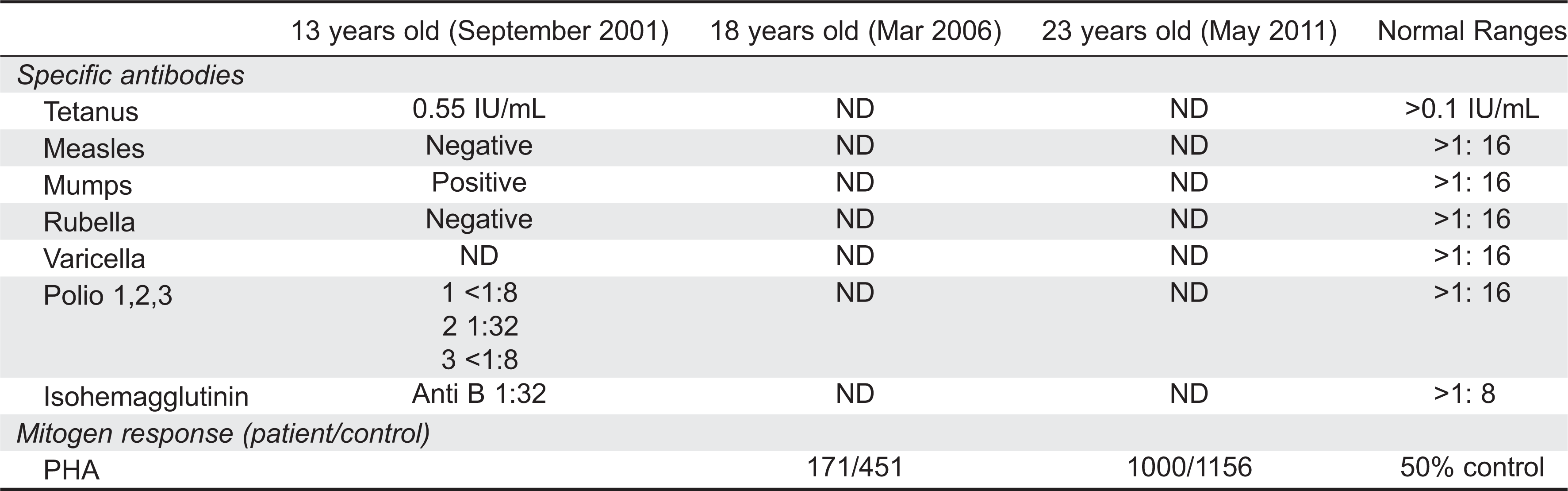
Figure 1:
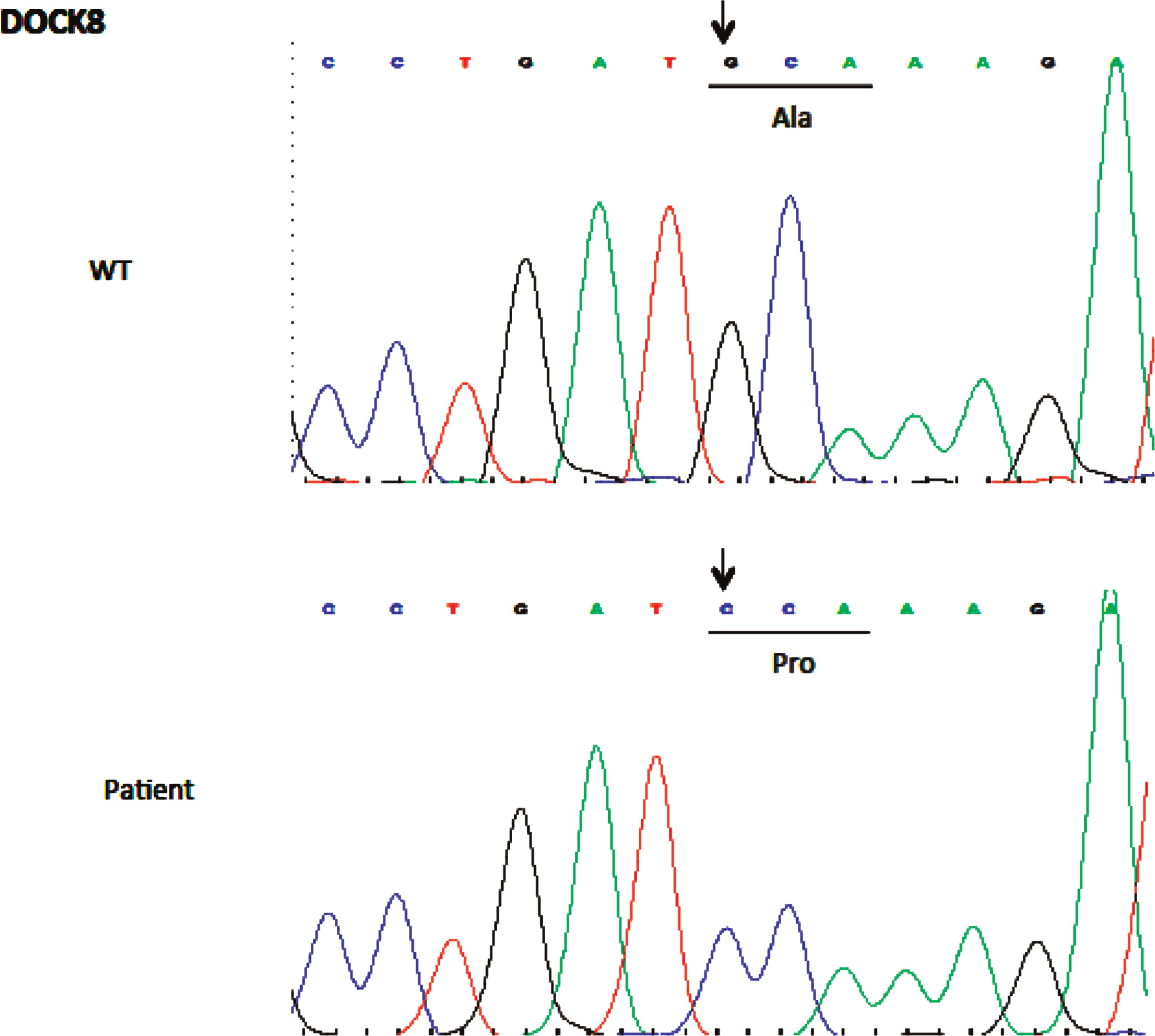
Figure 2:
Recurrence of colitis in a patient with chronic granulomatous disease associated colitis post hematopoietic stem cell transplantation
Arnon Broides
Pediatric Allergy Clinic, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel. [email protected]
Pediatric Allergy Clinic, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel. [email protected]
Chronic granulomatous disease (CGD) is a disorder of impaired reactive oxygen species production leading to inefficient intracellular killing. CGD patients typically suffer from recurrent life threatening infections and persistent non-infectious inflammation involving various organs including the gastrointestinal (GI) tract leading to colitis in many patients. Hematopoietic stem cell transplantations (HSCT) cure CGD and the associated inflammation leading to resolution of the associated colitis.
We present a CGD associated colitis patient in which the colitis recurred after HSCT.
The patient presented at the age of 19 months with one month of fever and diarrhea. Two liver abscesses were visualized by Ultrasound. Aspiration yielded pus, but no pathogen was found. He was diagnosed with CGD based on an abnormal dihydrorhodamine (DHR) assay. Empiric antimicrobial therapy was initiated with some improvement, and the patient was discharged. There was no significant personal medical history; however, his mother has ulcerative colitis. Six weeks later the patient was re-admitted due to continued intermittent fever and bloody diarrhea. Laboratory signs of inflammation were present. Gastrointestinal symptoms continued with intermittent bloody diarrhea. Imaging studies showed thickening of the colon. Endoscopies and biopsies also showed severe granulomatous inflammation. He was treated with mesalamine, itraconazole, antibiotics and interferon gamma. Although he received maximal therapy, he remained symptomatic. During this time, a mutation was found in the CYBB gene (R290X).
Hematopoietic stem cell transplantation was performed at 2 years of age using his HLA identical sister. The transplant was uneventful. Full donor engraftment was achieved at 17 days with normal DHR assay by day 24 with resolution of GI symptoms. However, the post HSCT course included prolonged asymptomatic EBV viremia, grade 2 skin and liver GVHD, Bronchiolitis obliterans and the colitis recurred at 28–38 months post HSCT necessitating multiple courses of steroids. Biopsies from Upper and lower GI showed non-specific severe inflammation. There were no granulomas to suggest reoccurrence of CGD or apoptotic bodies to suggest gut GVHD.
This unusual case shows recurrence of colitis in a CGD patient post HSCT without signs of CGD recurrence. It is possible that in this patient other genetic or environmental factors contributed to the recurrence of the colitis. However, it is also possible that in other reports of transplanted CGD associated colitis patients the colitis also recurred; however due to masking by immune suppression or lack of sufficient follow-up the recurrence was not recorded as such.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: e13 - e17
History
Version of record online: 29 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
2015. Immunodeficiency Canada Primary Immunodeficiency Video Conferences
Various dates, 2014
Multiple sites across Canada. LymphoSign Journal. 2(1): e13-e17. https://doi.org/10.14785/lpsn-2015-0903
Various dates, 2014
Multiple sites across Canada. LymphoSign Journal. 2(1): e13-e17. https://doi.org/10.14785/lpsn-2015-0903
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
