2014 Joint Immunodeficiency Canada/Israel Association of Allergy and Clinical Immunology Symposium
December 4, 2014
Dan Carmel Hotel
Haifa, Israel
TCR immunodeficiencies
José R. Regueiro
Immunology Dpt., Complutense University School of Medicine, 28040 Madrid, Spain
Immunology Dpt., Complutense University School of Medicine, 28040 Madrid, Spain
Introduction: The T-cell receptor complex (TCR) is an octameric transmembrane biosensor used by T cells to detect antigens. In humans it consists of a variable surface heterodimer (either αβ or γδ, Fig. 1) which actually binds antigens, and two different invariant CD3 heterodimers (CD3γɛ and CD3δɛ) and a single invariant CD247 homodimer (also termed ζζ), which transforms extracellular recognition into intracellular signals that regulate T-cell development, activation, and effector functions (Kuhns and Badgandi 2012).
Figure 1:
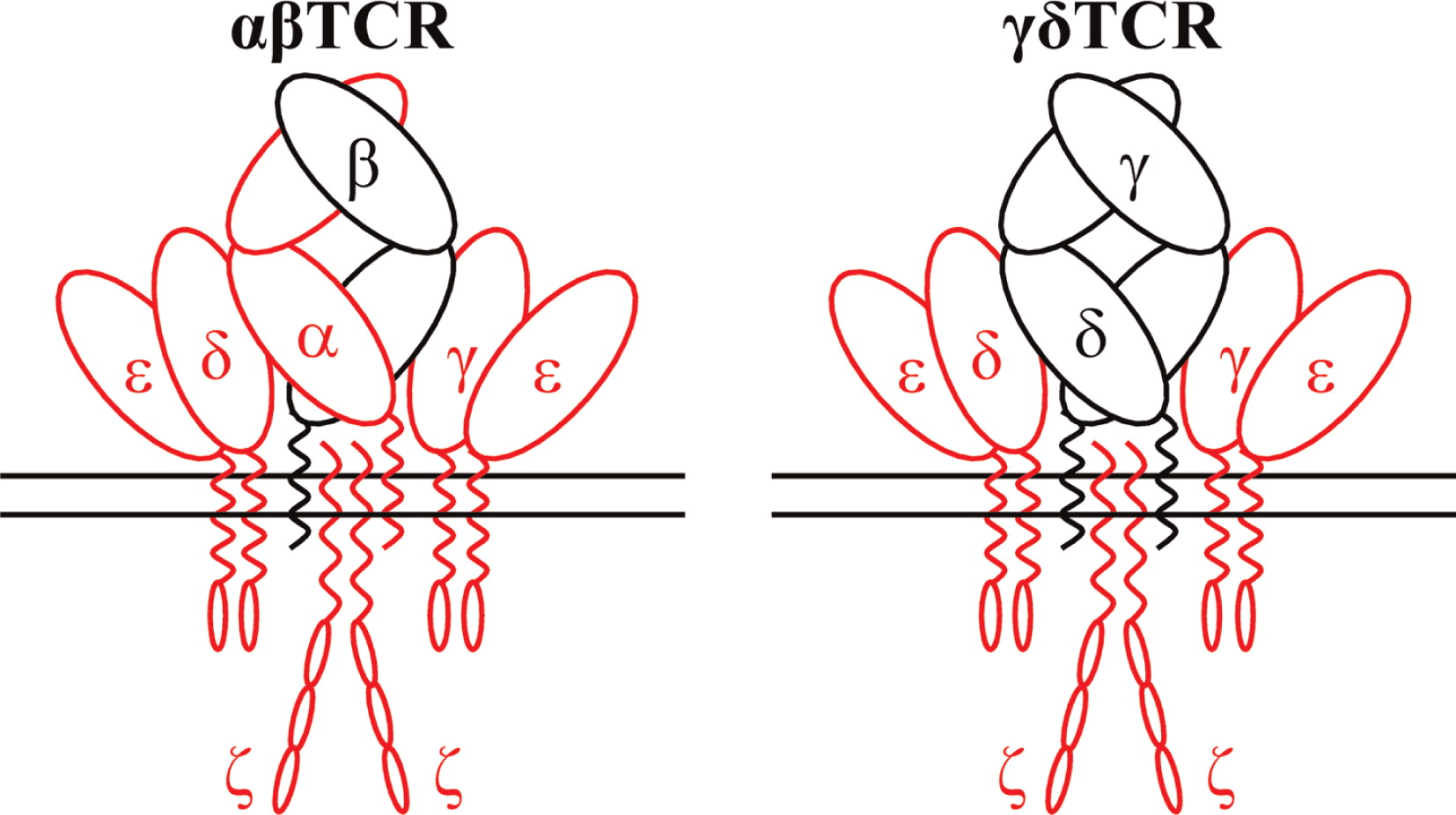
As with any other biological system, congenital defects in the TCR can cause immune dysfunctions. TCR immunodeficiencies (TCRID, Table 1) are very rare autosomal recessive diseases characterized by a prominent selective TCR complex expression defect frequently associated to peripheral blood T, but not B or NK, lymphocytopenia and SCID symptoms. They are caused by a range of severe or leaky mutations in the genes encoding for any of the TCR complex chains, although some notable exceptions remain, such as TCRβ, TCRγ and TCR. Small mutations in TRBC and TRGC (as described for TRAC) are unlikely because their constant regions gene segments are duplicated.
Table 1:
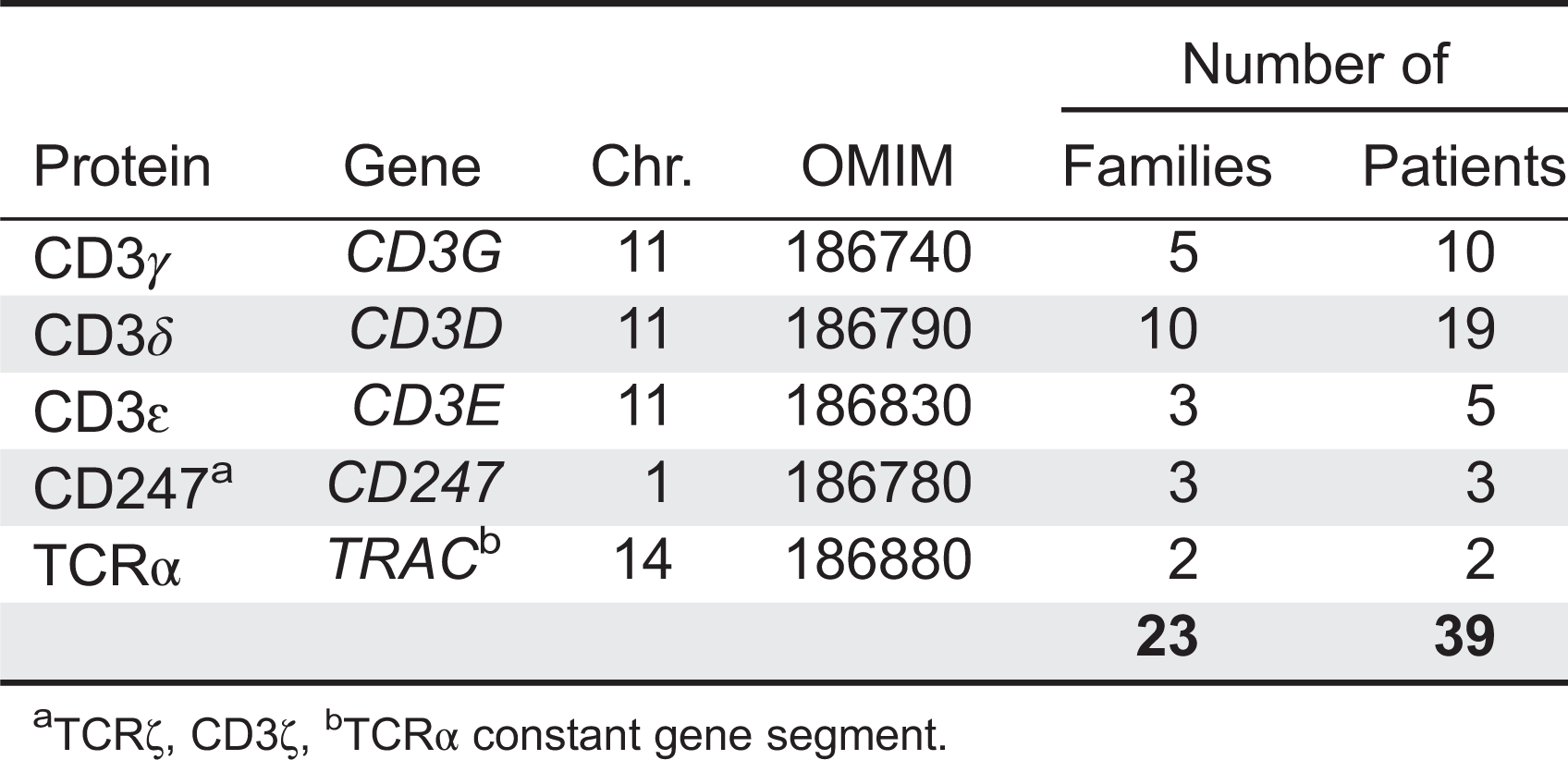
Diagnosis: Definitive: male or female patient with surface TCR complex expression defect, selective peripheral blood T lymphocytopenia (T-B+NK+ or T+/-B+NK+ phenotype) and mutations in a TCR complex gene (CD3G, CD3D, CD3E, CD247 or TRAC to date).
Probable: male or female patient with surface TCR expression defect and selective peripheral blood T lymphocytopenia (T-B+NK+ or T+/-B+NK+ phenotype).
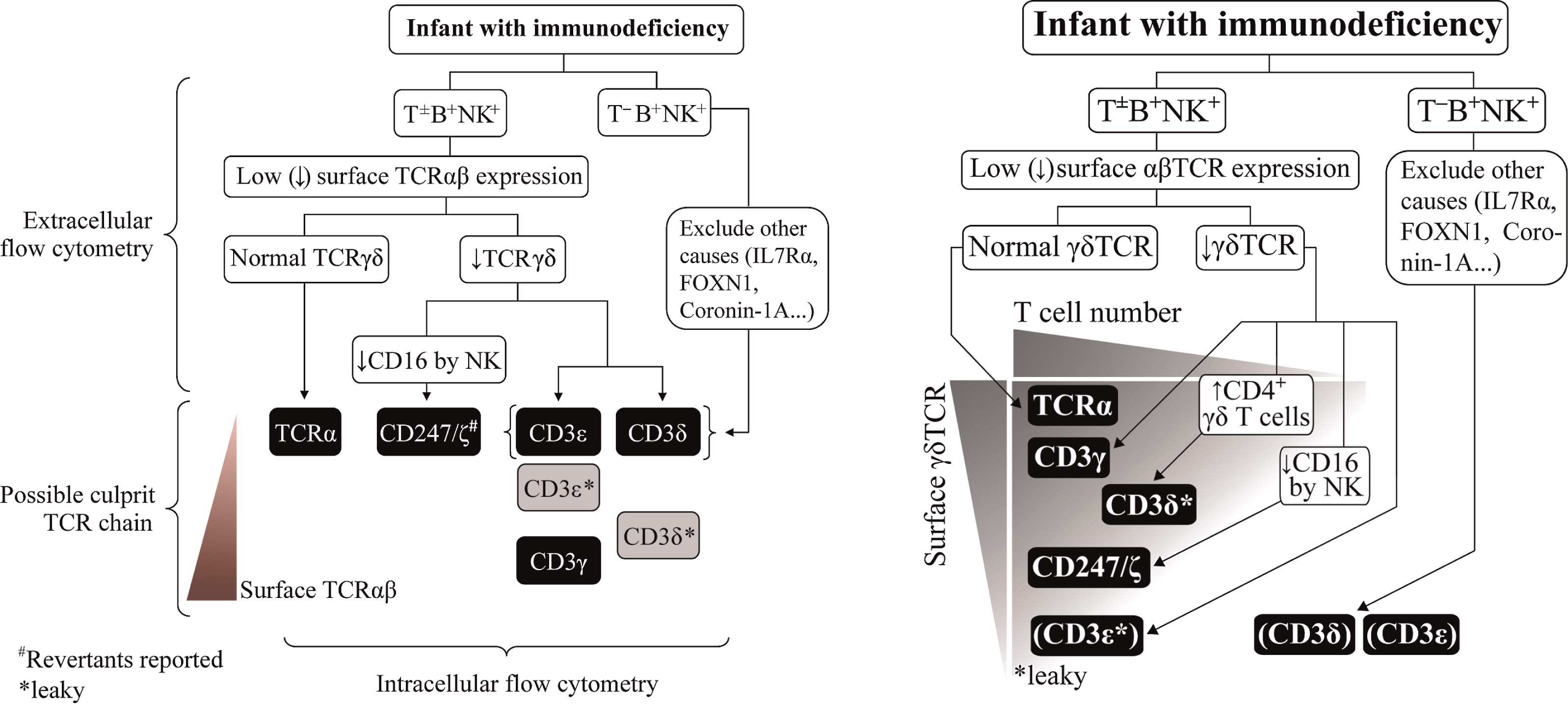
Spectrum of disease: from SCID (common) to healthy (rare, overlooked?). Complete CD3ɛ or CD3δ defects show a T-B+NK+ phenotype, whereas complete CD3γ or CD247 defects and partial defects (with some protein left) show the T+/-B+NK+ phenotype, including Tαβ-Tγδ+B+NK+. T cell revertants with normal TCR complex expression due to somatic mutations have been reported in CD247 deficiency.
Differential diagnosis: with patients showing T-B+NK+ or T+/-B+NK+ phenotypes, such as those with defects in IL7Rα, FOXN1, Coronin-1A, Zap70, MHC class I or II, PNP, ADA or DiGeorge syndrome.
A careful flow cytometry study can be helpful to diagnose TCRID (Fig. 2 left), particularly if γδ T cells are analyzed (Garcillán et al. 2014; Fig. 2 right).
Figure 2:
Treatment: Hematopoietic stem cell transplantation should be considered, the sooner the better (Marcus et al. 2011), even if mild ID is observed at presentation. Successfully transplanted patients have been shown to be healthy up to 18 years post-transplantation. Otherwise, the prognosis is very poor if SCID is present.
REFERENCES
Garcillán, B., Marín, A.V.M., Jiménez-Reinoso, A., Muñoz-Ruiz, M., García-León, M.J., Gil, J., Allende, L.M., Briones, A.C., Martínez-Naves, E., Toribio, M.L., and Regueiro, J.R. 2014. γδ T lymphocytes in the diagnosis of human T-cell receptor immunodeficiencies. Front Immunol.
Kuhns, M.S., and Badgandi, H.B. 2012. Piecing together the family portrait of TCR-CD3 complexes. Immunol. Rev. 250(1):120–143. doi: 10.1111/imr.12000.
Marin, A.V.M., Garcillán, B., Jiménez-Reinoso, A., Muñoz-Ruiz, M., Briones, A.C., Fernández-Malavé, E., Recio, M.J., and Regueiro, J.R. 2014. Human congenital T cell receptor disorders. Lymph. J.
Marcus, N., Takada, H., Law, J., Cowan, M.J., Gil, J., Regueiro, J.R., Plaza Lopez de Sabando, D., Lopez-Granados, E., Dalal, J., Friedrich, W., Manfred, H., Hanson, I.C., Grunebaum, E., Shearer, W.T., and Roifman, C.M. 2011. Hematopoietic stem cell transplantation for CD3delta deficiency. J. Allergy Clin. Immunol. 128(5): 1050–1057.
Recent advances in gene therapy approaches for the treatment of genetic disorders
Maria Pia Cicalese and Alessandro Aiuti
TIGET and Pediatric Immunohematology, San Raffaele Scientific Institute, Milano, Italy; Vita Salute University, Milano, Italy
TIGET and Pediatric Immunohematology, San Raffaele Scientific Institute, Milano, Italy; Vita Salute University, Milano, Italy
Gene therapy (GT) with hematopoietic stem cells (HSC) has been developing as a therapeutic strategy for the treatment of primary immunodeficiencies (PIDs) and other genetic diseases thanks to recent evidence of long-term disease correction as a result of ex vivo viral vector-mediated gene transfer into autologous hematopoietic stem cells.
The first results obtained with SCID-X1 using gamma-retroviral vectors showed T-cell reconstitution but 25% of treated patients developed a T-cell leukemia (Hacein-Bey-Abina et al. 2010). Modification of vector design using a self-inactivating retroviral vector has led to similar efficacy in the absence of serious adverse events. Gene therapy with retrovirally transduced HSC, combined to reduced intensity conditioning, has been developed as a successful and safe alternative strategy in ADA-SCID patients that do not have access to HLA-identical bone marrow transplantation (BMT) or for whom enzymatic replacement therapy (ERT) is not sufficient to maintain adequate immune reconstitution (Aiuti et al. 2009; Gaspar et al. 2011; Candotti et al. 2012). More than 40 ADA-SCID patients have been treated and none has developed leukemia or clonal proliferation. Retroviral vectors were then applied in clinical trials for patients with Wiskott-Aldrich Syndrome (WAS) and chronic granulomatous disease (CGD), the latter with transient benefit (Rivat et al. 2012). However, these trials were associated with high frequency of vector-related insertional mutagenic events (Cavazzana-Calvo et al. 2012; Braun et al. 2014).
Self-inactivating lentiviral vectors (LVs) represent a promising strategy with improved safety and efficacy. In the last few years, gene therapy trials for WAS patients based on LVs have started using different conditioning regimens and enrolling patients with severe clinical score and without a suitable BMT donor. Data from the first 3 WAS patients treated in GT clinical trial since 2010 at the San Raffaele Telethon Institute for Gene Therapy, Milan, have been recently reported (Aiuti et al. 2013). Our results show an improvement in the immune functions, an increase of the platelet counts with respect to the pre-GT values and normalization of the platelet volume, with clinical benefit for all treated patients, reduction of severity and frequency of infections in the absence of autoimmune manifestations (Aiuti et al. 2013).
The knowledge deriving from studies on molecular genetics, improved vector design and HSC biology are leading to preclinical development of GT for a wider spectrum of PIDs (Artemis deficiency, CD3γ deficiency, JAK3-SCID, LAD-1, PNP deficiency, RAG1/2 deficiency, IPEX, X-HIM, XLA, XLP, ZAP70 deficiency) (Passerini et al. 2013; Candotti 2014). Transition to the clinical phase will required a thorough evaluation of the GT potential risks and advantages over alternative treatments, such as allogeneic transplantation.
Apart from PIDs, in the last two decades significant advances have been made in GT for hemoglobinopathies and lysomal storage disorders. Clinical trials with β-/γ-globin lentivirus vectors are now open at multiple sites and transfusion independence following GT has been reported in one patient with β-thalassemia (Cavazzana-Calvo et al. 2010). Metabolic diseases have been another recent field of application of GT, with the proof of successful ABCD1 gene transfer to autologous hematopoietic stem cells by a lentiviral vector derived from HIV-1 in 3 patients with X-linked adrenoleukodystrophy (Cartier et al. 2009). Moreover, the first clinical trial of GT with lentiviral vector with ARSA gene showed extensive and stable ARSA gene replacement in the first 3 patients with metachromatic leukodystrophy and a lack of disease manifestation or progression in the treated patients several months beyond the predicted age of symptom onset (Biffi et al. 2013). In the current studies on the application of GT to other blood borne disorders, the use of different conditioning regimens, been identified as a crucial factor in achieving therapeutic levels of gene corrected, will have important implications for translating the experience of PID to diseases that require a higher therapeutic threshold.
If long-term efficacy and safety will be confirmed, gene therapy could become a standard treatment option for specific forms of PIDs and other monogenic diseases.
REFERENCES
Aiuti, A., Cattaneo, F., Galimberti, S., Benninghoff, U., Cassani, B., Callegaro, L., Scaramuzza, S., Andolfi, G., Mirolo, M., Brigida, I., Tabucchi, A., Carlucci, F., Eibl, M., Aker, M., Slavin, S., Al-Mousa, H., Al Ghonaium, A., Ferster, A., Duppenthaler, A., Notarangelo, L., Wintergerst, U., Buckley, R. H., Bregni, M., Marktel, S., Valsecchi, M. G., Rossi, P., Ciceri, F., Miniero, R., Bordignon, C., and Roncarolo, M.-G. 2009. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 360(5):447–458. PMID: 19179314. doi: 10.1056/NEJMoa0805817.
Aiuti, A., Biasco, L., Scaramuzza, S., Ferrua, F., Cicalese, M.P., Baricordi, C., Dionisio, F., Calabria, A., Giannelli, S., Castiello, M.C., Bosticardo, M., Evangelio, C., Assanelli, A., Casiraghi, M., Di Nunzio, S., Callegaro, L., Benati, C., Rizzardi, P., Pellin, D., Di Serio, C., Schmidt, M., Von Kalle, C., Gardner, J., Mehta, N., Neduva, V., Dow, D.J., Galy, A., Miniero, R., Finocchi, A., Metin, A., Banerjee, P.P., Orange, J.S., Galimberti, S., Valsecchi, M.G., Biffi, A., Montini, E., Villa, A., Ciceri, F., Roncarolo, M.G., and Naldini, L. 2013. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 341(6148):1233151. doi: 10.1126/science.1233151.
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., Baldoli, C., Martino, S., Calabria, A., Canale, S., Benedicenti, F., Vallanti, G., Biasco, L., Leo, S., Kabbara, N., Zanetti, G., Rizzo, W.B., Mehta, N.A.L., Cicalese, M.P., Casiraghi, M., Boelens, J.J., Del Carro, U., Dow, D.J., Schmidt, M., Assanelli, A., Neduva, V., Di Serio, C., Stupka, E., Gardner, J., Von Kalle, C., Bordignon, C., Ciceri, F., Rovelli, A., Roncarolo, M.G., Aiuti, A., Sessa, M., and Naldini, L. 2013. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 341(6148):1233158. doi: 10.1126/science.1233158.
Braun, C.J., Boztug, K., Paruzynski, A., Witzel, M., Schwarzer, A., Rothe, M., Modlich, U., Beier, R., Gohring, G., Steinemann, D., Fronza, R., Ball, C.R., Haemmerle, R., Naundorf, S., Kuhlcke, K., Rose, M., Fraser, C., Mathias, L., Ferrari, R., Abboud, M.R., Al-Herz, W., Kondratenko, I., Marodi, L., Glimm, H., Schlegelberger, B., Schambach, A., Albert, M.H., Schmidt, M., Von Kalle, C., and Klein, C. 2014. Gene therapy for Wiskott-Aldrich Syndrome—Long-Term efficacy and genotoxicity. Sci. Transl. Med. 6(227):227ra33. PMID: 24622513. doi: 10.1126/scitranslmed.3007280.
Candotti, F. 2014. Gene transfer into hematopoietic stem cells as treatment for primary immunodeficiency diseases. Int. J. Hematol. 99(4):383–392. doi: 10.1007/s12185-014-1524-z.
Candotti, F., Shaw, K.L., Muul, L., Carbonaro, D., Sokolic, R., Choi, C., Schurman, S.H., Garabedian, E., Kesserwan, C., Jagadeesh, G.J., Fu, P.-Y., Gschweng, E., Cooper, A., Tisdale, J.F., Weinberg, K.I., Crooks, G.M., Kapoor, N., Shah, A., Abdel-Azim, H., Yu, X.-J., Smogorzewska, M., Wayne, A.S., Rosenblatt, H.M., Davis, C.M., Hanson, C., Rishi, R.G., Wang, X., Gjertson, D., Yang, O.O., Balamurugan, A., Bauer, G., Ireland, J.A., Engel, B.C., Podsakoff, G.M., Hershfield, M.S., Blaese, R.M., Parkman, R., and Kohn, D.B. 2012. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 120:3635–3646. PMID: 22968453. doi: 10.1182/blood-2012-02-400937.
Cartier, N., Hacein-Bey-Abina, S., Bartholomae, C.C., Veres, G., Schmidt, M., Kutschera, I., Vidaud, M., Abel, U., Dal-Cortivo, L., Caccavelli, L., Mahlaoui, N., Kiermer, V., Mittelstaedt, D., Bellesme, C., Lahlou, N., Lefrere, F., Blanche, S., Audit, M., Payen, E., Leboulch, P., L'homme, B., Bougneres, P., Von Kalle, C., Fischer, A., Cavazzana-Calvo, M., and Aubourg, P. 2009. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 326(5954):818–823. doi: 10.1126/science.1171242.
Cavazzana-Calvo, M., Fischer, A., Hacein-Bey-Abina, S., Aiuti, A. 2012. Gene therapy for primary immunodeficiencies: Part 1. Curr. Opin. Immunol. 24(5):580–584. PMID: 22981681. doi: 10.1016/j.coi.2012.08.008.
Cavazzana-Calvo, M., Payen, E., Negre, O., Wang, G., Hehir, K., Fusil, F., Down, J., Denaro, M., Brady, T., Westerman, K., Cavallesco, R., Gillet-Legrand, B., Caccavelli, L., Sgarra, R., Maouche-Chrétien, L., Bernaudin, F., Girot, R., Dorazio, R., Mulder, G.-J., Polack, A., Bank, A., Soulier, J., Larghero, J., Kabbara, N., Dalle, B., Gourmel, B., Socie, G., Chrétien, S., Cartier, N., Aubourg, P., Fischer, A., Cornetta, K., Galacteros, F., Beuzard, Y., Gluckman, E., Bushman, F., Hacein-Bey-Abina, S., and Leboulch, P. 2010. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 467(7313):318–322. doi: 10.1038/nature09328.
Gaspar, H.B., Cooray, S., Gilmour, K.C., Parsley, K.L., Adams, S., Howe, S.J., Al Ghonaium, A., Bayford, J., Brown, L., Davies, E.G., Kinnon, C., Thrasher, A.J. 2011. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 3(97):97ra79. PMID: 21865537.
Hacein-Bey-Abina, S., Hauer, J., Lim, A., Picard, C., Wang, G.P., Berry, C.C., Martinache, C., Rieux-Laucat, F., Latour, S., Belohradsky, B.H., Leiva, L., Sorensen, R., Debré, M., Casanova, J.L., Blanche, S., Durandy, A., Bushman, F.D., Fischer, A., and Cavazzana-Calvo, M. 2010. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 363(4):355–364. doi: 10.1056/NEJMoa1000164.
Passerini, L., Mel, E.R., Sartirana, C., Fousteri, G., Bondanza, A., Naldini, L., Roncarolo, M.G., and Bacchetta, R. 2013. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci. Transl. Med. 5(215):215ra174. doi: 10.1126/scitranslmed.3007320.
Rivat, C., Santilli, G., Gaspar, H.B., Thrasher, A.J. 2012. Gene therapy for primary immunodeficiencies. Hum. Gene. Ther. 23(7):668–675. PMID: 22691036. doi: 10.1089/hum.2012.116.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 1 • March 2015
Pages: e9 - e12
History
Version of record online: 29 October 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
2015. 2014 Joint Immunodeficiency Canada/Israel Association of Allergy and Clinical Immunology Symposium
December 4, 2014
Dan Carmel Hotel
Haifa, Israel. LymphoSign Journal. 2(1): e9-e12. https://doi.org/10.14785/lpsn-2015-0902
December 4, 2014
Dan Carmel Hotel
Haifa, Israel. LymphoSign Journal. 2(1): e9-e12. https://doi.org/10.14785/lpsn-2015-0902
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
