Hemophagocytic lymphohistiocytosis in a patient with CD3δ deficiency
Abstract
Introduction: Primary hemophagocytic lymphohistiocytosis (HLH) is a life-threatening inflammatory process that has been linked to abnormal cytotoxic T-cell and natural killer (NK) cell function. We report on the first case of severe combined immunodeficiency (SCID) caused by a CD3δ mutation presenting with HLH in a female of Mennonite descent.
Case Description: A Low-German-speaking Mennonite female with past medical history of eczema, mouth sores, and refractory oral and diaper candidiasis presented at the age of 6 months with vomiting, diarrhea, and lethargy. The patient developed HLH that was refractory to treatment and led to multi-organ failure. Immunological evaluation was diagnostic for SCID and post-mortem genetic testing confirmed a homozygous mutation in CD3δ that was previously described in Mennonites.
Method: Targeted molecular testing for CD3δ deficiency confirmed a homozygous C-to-T transition at nucleotide position 202, predicting a premature stop codon, with a truncation at residue 68 (R68X) in the extracellular domain of the protein.
Discussion: Many primary immunodeficiency diseases (PID) that affect cytotoxic T cells and NK cells have presented with HLH. However, a growing number of PID with no obvious NK-cell defect have also been found to predispose patients to HLH, suggesting that failure of NK activity is not the only mechanism leading to this unusual form of inflammation.
Conclusion: CD3δ is known to be critical for T-cell but not NK-cell development, which may suggest an alternate mechanism for overwhelming inflammation leading to HLH.
Statement of novelty: This is the first case report of CD3δ deficiency presenting with HLH.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening inflammatory process with a poorly understood pathogenesis (Janka and Lehmberg 2014; Risma and Jordan 2012). Because the condition is associated with multiple different disorders, the Histiocyte Society has classified cases into 2 groups: primary (familial) HLH, and secondary (acquired) HLH.
Secondary HLH appears in association with infections (Epstein–Barr virus (EBV), Cytomegalovirus (CMV)), malignancy, arthritis, metabolic disorders, or as a consequence of drug toxicity (Henter et al. 1991; Janka et al. 1998).
Primary HLH typically presents in young infants who may have a history of other affected siblings or relatives. Several genetic defects have been identified to predispose infants to HLH, all of which are involved in cytotoxic T-cell and natural killer (NK) cell function. In North America, the most frequent mutations are in the PFR1 gene that encodes for Perforin 1. Other genes including UNC13D and STX11 whose products are involved in cytotoxic granule trafficking and exocytosis, respectively, have been implicated (Zhang 2011). In addition, cases of primary immunodeficiency (PID) such as X-linked severe combined immunodeficiency (SCID) (Grunebaum and Roifman 2002), Chediak–Higashi syndrome, and Griscelli syndrome type 2, can also present with HLH. Abnormal cytotoxic function is common to these disorders and may explain, at least in part, the association with HLH. In contrast, a growing number of PID with no obvious NK-cell defect have also been found to predispose patients to HLH, suggesting that failure of NK activity is not the only mechanism leading to this unusual form of inflammation. Wiskott–Aldrich syndrome, DiGeorge syndrome (primarily the 22q11.2 microdeletion syndrome), and STAT1 deficiency are prime examples (Introne et al. 1999; Menasche et al. 2000; Cesaro et al. 2003; Pasic et al. 2003; Faitelson and Grunebaum 2014). It is possible that the lack of cytotoxic T-cell function might play a role in this predisposition.
The symptoms and signs of HLH are nonspecific including fever, hepatosplenomegaly, lymphadenopathy, cytopenia, seizures, and encephalopathic changes (Henter et al. 2007) Without treatment, less than 5% of diagnosed patients survive (Janka 1983). In spite of major advances made in treating this condition, mortality remains high, even in treated individuals. Treatment is based on the attempt to dampen inflammation with immunosuppressive and apoptosis-inducing agents such as dexamethasone, etoposide, and cyclosporine, but in some cases, bone marrow transplantation cannot be avoided. We report the first case of SCID caused by a CD3δ mutation presenting with HLH.
Patient report
An unimmunized Low-German-speaking Mennonite female, born to a consanguineous couple, presented at the age of 6 months with nonbilious emesis, diarrhea, and lethargy with a 1 month history of fever, nonproductive cough, and 1 kg weight loss. She was treated initially with 2 courses of amoxicillin for a presumed diagnosis of pneumonia, with no improvement. Her past medical history was remarkable for eczema, recurrent mouth sores, and refractory oral and diaper candidiasis from 4 months of age, despite appropriate treatment. Older siblings were unwell, with symptoms consistent with a viral upper respiratory tract infection the week prior to presentation. Upon admission to hospital a petechial rash, hepatomegaly, and hypotonia were noticed on the physical exam.
Investigations revealed anemia (Hgb 88 g/L), thrombocytopenia (118 × 109/L), leukopenia (WBC 4 × 109/L), and differential counts showing neutropenia (0.70 × 109/L) and lymphopenia (2.15 × 109/L, Table 1). Moreover, she had a very high ferritin (23 674 µg/L), elevated triglyceride (12.24 mmol/L), hypofibrinogenemia (1.37 g/L), coagulopathy (INR 1.8, aPTT 64 s), and transaminitis (ALT 303 U/L, AST 2145 U/L). The soluble CD163 was elevated (2270 ng/mL) as well as the soluble IL-2 receptor (4683 U/mL). A nasopharyngeal swab was positive for adenovirus, and a concentrated urine sample (specific gravity 1.025) demonstrated nitrites, blood, and trace ketones; it later grew Klebsiella pneumoniae on culture. Thymic tissue was notably absent on a chest radiograph, and a CT chest scan revealed multiple infiltrates suspicious for fungus. CMV and EBV polymerase chain reaction (PCR) analyses on blood were negative. MRI of the brain performed for fluctuating level of consciousness and unilateral facial droop revealed left cerebellar hemispheric calcification suspicious for previous hemorrhage. Cerebrospinal fluid showed elevated protein without pleocytosis.
Table 1:
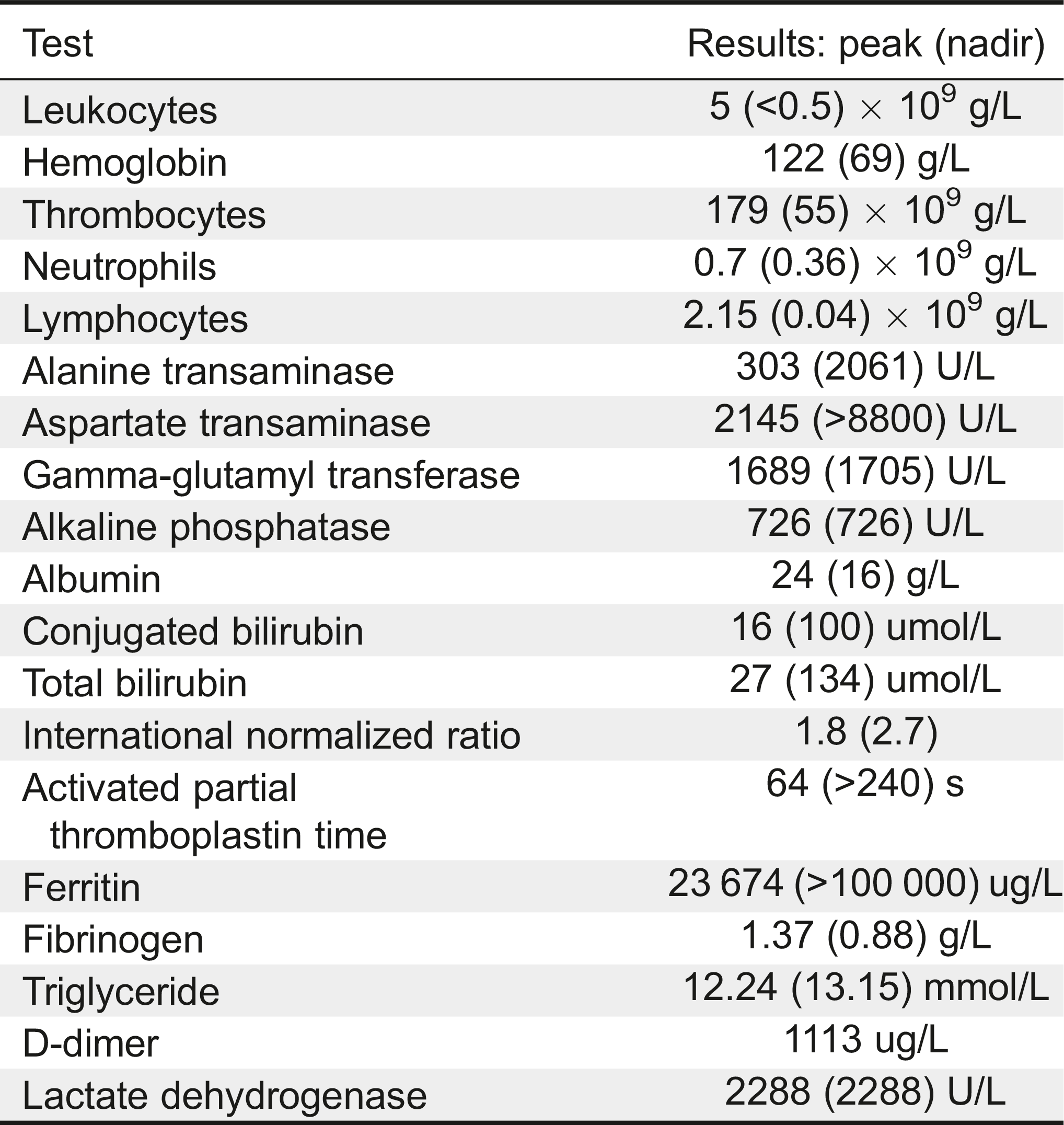
Dexamethasone was started for a presumptive diagnosis of HLH. Peripheral blood lymphocyte subsets numbers displayed very low CD3+ (38 cells/µL), low CD4+ (24 cells/µL), low CD8+ (1 cell/µL), normal CD19+ (1071 cells/µL), and low CD16+56+ (79 cells/µL). Additional immunological work-up included normal IgG (4.8 g/L), elevated IgA (1.3 g/L), normal IgM (0.40 g/L), and undetectable IgE (<2 kU/L). The patient was not immunized, hence, specific antibodies were not done.
A bone marrow aspirate, which was done within 5 days of initiating the treatment, did not show morphologic hemophagocytosis. Flow cytometry of the sample showed very low T-cell numbers with only 1% of the lymphocyte population.
A diagnosis of SCID was established. Despite intensive treatment with broad-spectrum antibiotics, IVIG, corticosteroids, and etoposide, the patient's condition deteriorated. She required multiple doses of vitamin K, factor VIIa, and transexamic acid; a pantoprazole infusion; and transfusions of frozen plasma, cryoprecipitate, packed red blood cells, and platelets, as she developed medically refractory hemorrhage involving the upper and lower GI tract and vascular access lines. She developed intractable HLH (ferritin >100 000 µg/L) with multi-organ failure (liver failure, encephalopathy, and respiratory failure) and refractory bleeding. She was cared for by multiple services including hematology/oncology, infectious diseases, neurology, gastroenterology, cardiology, genetics, general surgery, anesthesia, as well as the multidisciplinary pediatric critical care unit. HLA typing with planned search for a suitable stem cell donor was initiated. However, with the ongoing HLH, refractory bleeding, and encephalopathy, the family opted not to continue the administration of blood products and chose to have palliative care. The patient died within 2 weeks of hospitalization.
Molecular testing revealed a homozygous mutation in the CD3δ gene that had been previously described in the Low-German-speaking Mennonite population (Dadi et al. 2003).
Methods
Genomic DNA was amplified by PCR with specific primers for the coding sequence of CD3δ exon 2 and the flanking intronic regions (primers sequenced as previously published by Dadi et al. (2003)). Products were sequenced with the GenomeLab Dye Terminatorcycle Sequencing Quick Start Kit (Beckman Coulter, Brea, California) and analyzed on a CEQ 8000 Genetic Analysis System (Beckman Coulter, Brea, California). Targeted molecular testing for CD3δ deficiency confirmed a homozygous C-to-T transition at nucleotide position 202, predicting a premature stop codon, with a truncation at residue 68 (R68X) in the extracellular domain of the protein. This mutation has previously been reported in the Low-German-speaking Mennonite population (Dadi et al. 2003). Fluorescence in situ hybridization for the 22q11.2 microdeletion yielded normal results. Targeted perforin mutation testing for a known Old Order Mennonite mutation was negative.
Discussion
CD3δ deficiency was first described by Dadi et al. (2003), in infants of Low-German-speaking Mennonite descent. These patients presented with typical features of SCID including oral thrush, failure to thrive, and lung infections. Evaluation of their immune system revealed a complete lack of circulating mature CD4+ and CD8+ T cells. However, B-cell and NK-cell numbers were normal. Subsequently, other mutations in CD3δ have been identified in Japanese and French SCID patients, again presenting in an identical fashion (de Saint Basile et al. 2004; Takada et al. 2005). More recently, 2 unrelated Low-German-speaking Mennonite patients with homozygous mutations in CD3δ were described in Western Canada (Lam et al. 2014). These patients presented with respiratory distress and bronchial aspirate detected Pneumocystis jiroveci at less than 4 months of age. One had a similar history to our patient with persistent thrush and failure to thrive, and both had a history of fevers and respiratory infections. Flow cytometry in these patients revealed absent T cells with normal B cells and normal NK cells. One child received a sibling-matched hematopoetic stem cell transplant but died 2 months post-transplant due to metapneumovirus infection. The second child died at 3.4 months of age with worsening respiratory status. Neither child had obvious features of HLH reported. A leaky type of CD3δ deficiency has been discovered in Ecuadorian children (Gil et al. 2011). Unlike the original cases, these patients had normal numbers of circulating TCRγδ+ cells but reduced TCRαβ+ cells. In these cases NK-cell function was tested and as expected was found to be normal. Recently, patients with CD3δ deficiency have been reported to present with Omenn syndrome, another type of aberrant inflammatory disorder associated with SCID. Omenn Syndrome typically presents with severe erythroderma, hepatosplenomegaly, and increased eosinophilia (Villa et al. 2008). This abnormal process is believed to emerge from loss of immune surveillance and control by some harmful expanding autologous T-cell clones. Immunosuppression is the hallmark of treatment in this disorder, similar to HLH. Indeed, HLH has been reported in a growing number of T-cell PID in which normal immune surveillance is lost, such as X-linked SCID (Grunebaum and Roifman 2002), Zap70 deficiency, and others. A recent published paper has reviewed the association between HLH and PID (Faitelson and Grunebaum 2014).
We describe the first case of HLH in a patient with CD3δ deficiency. Genetic analysis of PFR1, UNC13D, and STX11 were normal in this patient. Although the patient's history of eczema, recurrent mouth sores, and refractory oral and diaper candidiasis were suspicious for an underlying immunodeficiency, the initial lymphocyte phenotype showed somewhat reduced (borderline) numbers of NK cells, not typical of SCID conditions known in the Mennonite population (i.e., ADA, ZAP70 deficiencies). Both CD3δ deficiency and CD3γ deficiency typically have a lymphocyte phenotype of T–B+NK+; however, in the current case it is possible that treatment with dexamethasone led to the reduction in NK cells. Owing to the previous CD3δ cases reported in the Mennonite community, we explored this diagnostic possibility first. This highlights the importance of taking into account the ethnic and genetic background of patients and testing for the known SCID mutations common to ethnicity. This approach is both time saving and cost saving compared with pursuing a large panel of immunodeficiency genes or exome sequencing. Indeed in this case, ADA deficiency (T–B–NK–) or Zap70 deficiency (T+B+NK+), which are common in this ethnic group, were not tested because of their different immunophenotype (International Union of Immunological Societies Expert Committee on Primary et al. 2009; Kwan et al. 2014).
The knowledge of population-specific SCID mutations becomes increasingly important in light of the introduction of provincial newborn screening for SCID. Newborn screening for SCID started in Ontario in September of 2013, 5 months after the birth of our reported patient. Newborn screening for SCID is a quantitative PCR analysis of the T-cell receptor excision circles (TRECs). TRECs are a normal by-product of the T-cell maturation process when the T-cell receptor gene undergoes rearrangements to produce the final unique T-cell receptor. Patients with SCID typically have a very low number of T cells and therefore a low number of TRECs. Although newborn screening for SCID was not yet in place when our patient was born, we were able to retrieve the neonatal blood dot card and confirmed the absence of TRECs. The early management of this child would have been different if she had been identified as a neonate to have SCID using newborn screening. This also raises the potential value of preconceptual carrier testing for the known SCID mutations in the Low-German-speaking Mennonite population as a means of identifying couples at an increased risk to have a child with SCID. This could allow for the option of prenatal and (or) postnatal molecular diagnosis.
This paper illustrates that HLH may occur in patients with CD3δ deficiency, likely due to the lack of functional cytotoxic T cells. Although the precise mechanism leading to HLH in CD3δ deficiency remains to be determined, it is plausible to assume that the absence of cytotoxic T cells, even in the presence of normal NK cells, may be a critical predisposing factor (George 2014). This has also been observed in some case reports of PID that are not typically affecting the NK cell number or function (Introne et al. 1999; Menasche et al. 2000; Cesaro et al. 2003; Pasic et al. 2003; Faitelson and Grunebaum 2014). It might be related, at least partially, to the lack of defense mechanisms against infectious triggers known to predispose to HLH (Faitelson and Grunebaum 2014). On the other hand, in mouse models, the presence of cytotoxic T cells in perforin knock-out mice infected with lymphocytic choriomeningitic virus lead to HLH. Depleting the cytotoxic T cells ablated the development of HLH, suggesting that the presence of overactive cytotoxic T cells plays a role in its pathogenesis (Jordan et al. 2004). Alternatively, more extensive genetic studies such as whole genome sequencing might reveal whether other modifier genes could have contributed to the development of HLH in this patient.
Acknowledgment
DNA sequence analysis was done at The Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency.
REFERENCES
Cesaro S., Messina C., Sainati L., Danesino C., and Arico M. Del 22Q11.2 and hemophagocytic lymphohistiocytosis: A non-random association Am. J. Med. Genet. A 2003 116A 2 208 -209
Dadi H.K., Simon A.J., and Roifman C.M. Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency N. Engl. J. Med 2003 349 19 1821 -1828
de Saint Basile G., Geissmann F., Flori E., Uring-Lambert B., Soudais C., Cavazzana-Calvo M., Durandy A., Jabado N., Fischer A., and Le Deist F. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3 J Clin Invest 2004 114 10 1512 -1517
Faitelson Y. and Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders Clin. Immunol 2014 155 1 118 -125
George M.R. Hemophagocytic lymphohistiocytosis: Review of etiologies and management J. Blood Med 2014 5 69 -86
Gil J., Busto E.M., Garcillan B., Chean C., Garcia-Rodriguez M.C., Diaz-Alderete A., Navarro J., Reine J., Mencia A., Gurbindo D., Belendez C., Gordillo I., Duchniewicz M., Hohne K., Garcia-Sanchez F., Fernandez-Cruz E., Lopez-Granados E., Schamel W.W., Moreno-Pelayo M.A., Recio M.J., and Regueiro J.R. A leaky mutation in CD3D differentially affects alphabeta and gammadelta T cells and leads to a Talphabeta-Tgammadelta+B+NK+ human SCID J. Clin. Invest 2011 121 10 3872 -3876
Grunebaum E. and Roifman C.M. Gene abnormalities in patients with hemophagocytic lymphohistiocytosis Isr. Med. Assoc. J 2002 4 5 366 -369
Henter J.I., Elinder G., and Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society Semin. Oncol 1991 18 1 29 -33
Henter J.I., Horne A., Arico M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., McClain K., Webb D., Winiarski J., and Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatr. Blood Cancer 2007 48 2 124 -131
International Union of Immunological Societies Expert Committee on Primary I., Notarangelo L.D., Fischer A., Geha R. S., Casanova J.L., Chapel H., Conley M.E., Cunningham-Rundles C., Etzioni A., Hammartrom L., Nonoyama S., Ochs H.D., Puck J., Roifman C., Seger R., and Wedgwood J. Primary immunodeficiencies: 2009 update J. Allergy Clin. Immunol 2009 124 6 1161 -1178
Introne W., Boissy R.E., and Gahl W.A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome Mol. Genet. Metab 1999 68 2 283 -303
Janka G., Imashuku S., Elinder G., Schneider M., and Henter J.I. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis Hematol. Oncol. Clin. North Am 1998 12 2 435 -444
Janka G.E. Familial hemophagocytic lymphohistiocytosis Eur. J. Pediatr 1983 140 3 221 -230
Janka G.E. and Lehmberg K. Hemophagocytic syndromes–An update Blood Rev 2014 28 4 135 -142
Jordan M.B., Hildeman D., Kappler J., and Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder Blood 2004 104 3 735 -743
Kwan A., Abraham R.S., Currier R., Brower A., Andruszewski K., Abbott J.K., Baker M., Ballow M., Bartoshesky L.E., Bonilla F.A., Brokopp C., Brooks E., Caggana M., Celestin J., Church J.A., Comeau A.M., Connelly J.A., Cowan M.J., Cunningham-Rundles C., Dasu T., Dave N., De La Morena M.T., Duffner U., Fong C.T., Forbes L., Freedenberg D., Gelfand E.W., Hale J.E., Hanson I.C., Hay B.N., Hu D., Infante A., Johnson D., Kapoor N., Kay D.M., Kohn D.B., Lee R., Lehman H., Lin Z., Lorey F., Abdel-Mageed A., Manning A., McGhee S., Moore T.B., Naides S.J., Notarangelo L.D., Orange J.S., Pai S.Y., Porteus M., Rodriguez R., Romberg N., Routes J., Ruehle M., Rubenstein A., Saavedra-Matiz C.A., Scott G., Scott P.M., Secord E., Seroogy C., Shearer W.T., Siegel S., Silvers S.K., Stiehm E.R., Sugerman R.W., Sullivan J.L., Tanksley S., Tierce M.L., Verbsky J., Vogel B., Walker R., Walkovich K., Walter J.E., Wasserman R.L., Watson M.S., Weinberg G.A., Weiner L.B., Wood H., Yates A.B., Puck J.M., and Bonagura V.R. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States JAMA 2014 312 7 729 -738
Lam S., Kavadas F.D., Haider S., and Noseworthy M.E. The respiratory presentation of severe combined immunodeficiency in two Mennonite children at a tertiary centre highlighting the importance of recognizing this pediatric emergency Can. Respir. J 2014 21 1 17 -19
Menasche G., Pastural E., Feldmann J., Certain S., Ersoy F., Dupuis S., Wulffraat N., Bianchi D., Fischer A., Le Deist F., and de Saint Basile G. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome Nat. Genet 2000 25 2 173 -176
Pasic S., Micic D., and Kuzmanovic M. Epstein–Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott–Aldrich syndrome Acta. Paediatr 2003 92 7 859 -861
Risma K. and Jordan M.B. Hemophagocytic lymphohistiocytosis: Updates and evolving concepts Curr. Opin. Pediatr 2012 24 1 9 -15
Takada H., Nomura A., Roifman C.M., and Hara T. Severe combined immunodeficiency caused by a splicing abnormality of the CD3delta gene Eur. J. Pediatr 2005 164 5 311 -314
Villa A., Notarangelo L.D., and Roifman C.M. Omenn syndrome: Inflammation in leaky severe combined immunodeficiency J. Allergy Clin. Immunol 2008 122 6 1082 -1086
Zhang K., Jordan M.B., Marsh R.A., Johnson J.A., Kissell D., Meller J., Villanueva J., Risma K.A., Wei Q., Klein P.S., and Filipovich A.H. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH Blood 2011 118 22 5794 -5798
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 4 • December 2015
Pages: 201 - 206
History
Received: 22 April 2015
Accepted: 26 August 2015
Accepted manuscript online: 27 August 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Mohammad Alsalamah, Amrita Sarpal, Victoria Mok Siu, Paul Gibson, CA Rupar, Michelle Barton, Marina I Salvadori, and Sharan Goobie. 2015. Hemophagocytic lymphohistiocytosis in a patient with CD3δ deficiency. LymphoSign Journal.
2(4): 201-206. https://doi.org/10.14785/lpsn-2015-0006
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
