Atypical hemolytic-uremic syndrome in a patient with adenosine deaminase deficiency
Abstract
Inherited defects in the ubiquitous adenosine deaminase (ADA) enzyme disrupt the function of the immune system as well as many other organs and tissues. Some patients may also suffer from kidney damage. Here we report on an ADA-deficient patient who was treated with ADA replacement therapy from infancy and at 6 years of age developed acute kidney failure, thrombocytopenia, and severe anemia. A kidney biopsy demonstrated mesangiolysis and occlusion of kidney loops by erythrocytes and platelet aggregates, which is consistent with hemolytic-uremic syndrome (HUS). There was no evidence of exposure to Shiga toxins, nor were any complement abnormalities detected. The kidney function improved following hemodialysis. Our report demonstrates the increased susceptibility of ADA-deficient patients to develop HUS and expands the nonimmune abnormalities associated with ADA deficiency. This further emphasizes the vigilance required when caring for such patients.
Statement of novelty: Here we provide the first detailed clinical and histological characterization of hemolytic-uremic syndrome developing in an ADA-deficient patient.
Introduction
Adenosine deaminase (ADA) is a ubiquitous enzyme that catalyzes the deamination of adenosine and deoxyadenosine to inosine and 2′-deoxyinosine, respectively. Inherited defects that disrupt ADA enzyme activity lead to marked lymphopenia, resulting in severe immune deficiency with increased susceptibility to bacterial, viral, and opportunistic infections, typically in infancy (Grunebaum et al. 2013). ADA-deficient patients may also develop various autoimmune features, including autoimmune cytopenia, possibly due to abnormalities in regulatory T cells (Sauer et al. 2012). Treatment options for ADA deficiency include hematopoietic stem cell transplantation (HSCT), preferably from a human leukocyte antigen (HLA)-identical sibling donor. In the absence of an appropriate donor for HSCT, experimental gene therapy or repeated injections of polyethylene glycol conjugated-ADA (PEG-ADA) can be used (Gaspar et al. 2009). Long-term survival with ADA enzyme replacement is possible, although many patients remain susceptible to infections, autoimmunity, and malignancy (Husain et al. 2007). ADA is ubiquitously expressed; therefore, it is not surprising that impaired ADA activity also disrupts many other cells, tissues, and organs including hepatocytes, chondrocytes, osteoblasts, alveolar macrophages, etc. (Bollinger et al. 1996; Grunebaum et al. 2012). ADA deficiency can also affect patients’ bone marrow (Sokolic et al. 2011) and kidneys (Ratech et al. 1985).
Hemolytic-uremic syndrome (HUS) is the most common cause of acute kidney failure in children, affecting 0.2–4.2 children per 100 000 children worldwide. HUS is characterized by progressive kidney failure, nonimmune (Coombs-negative) microangiopathic hemolytic anemia, and thrombocytopenia. HUS is most often associated with a Shiga-like toxin produced by entero-hemorrhagic E. coli O157:H7. Rarer cases have also been known to be triggered by neuraminidase-producing Streptoccus pneumoniae (Brandt et al. 2002), although HUS unrelated to toxins, often categorized as atypical HUS, is being recognized at an increasing frequency (Keir and Coward 2011). Kidney biopsies from patients suffering from HUS often show vascular wall thickening as well as swelling and detachment of the endothelial cells from the basement membrane. Additionally, there is an accumulation of material in the subendothelial space, intraluminal platelet thrombosis, partial or complete vessel luminal obstruction, and fragmentation of red blood cells (Keir and Coward 2011). Here we describe an ADA-deficient patient who developed atypical HUS.
Case report
The Institutional Research Ethics Board at The Hospital for Sick Children, Toronto, approved the study and a written informed consent for the research was obtained. The female patient, born to Inuit nonconsanguineous parents, was identified in infancy as suffering from ADA deficiency based on absent ADA activity in the erythrocytes and the identification of compound heterozygous mutations in the ADA gene (c.424C> T and c.955_959delGAAGA). An HLA-matched donor for HSCT could not be found; therefore, the patient was treated with PEG-ADA (ADAGEN®) resulting in a very limited immune reconstitution (Table 1). The patient also received prophylactic sulfamethoxazole/thrimethoprim and monthly intravenous immunoglobulin (IVIG) infusions without significant infections or autoimmunity. Sensorineural hearing loss was noted in infancy and stage I hypertension, that didn't require treatment, was observed in early childhood. Kidney function and ultrasound were reported to be normal.
Table 1:
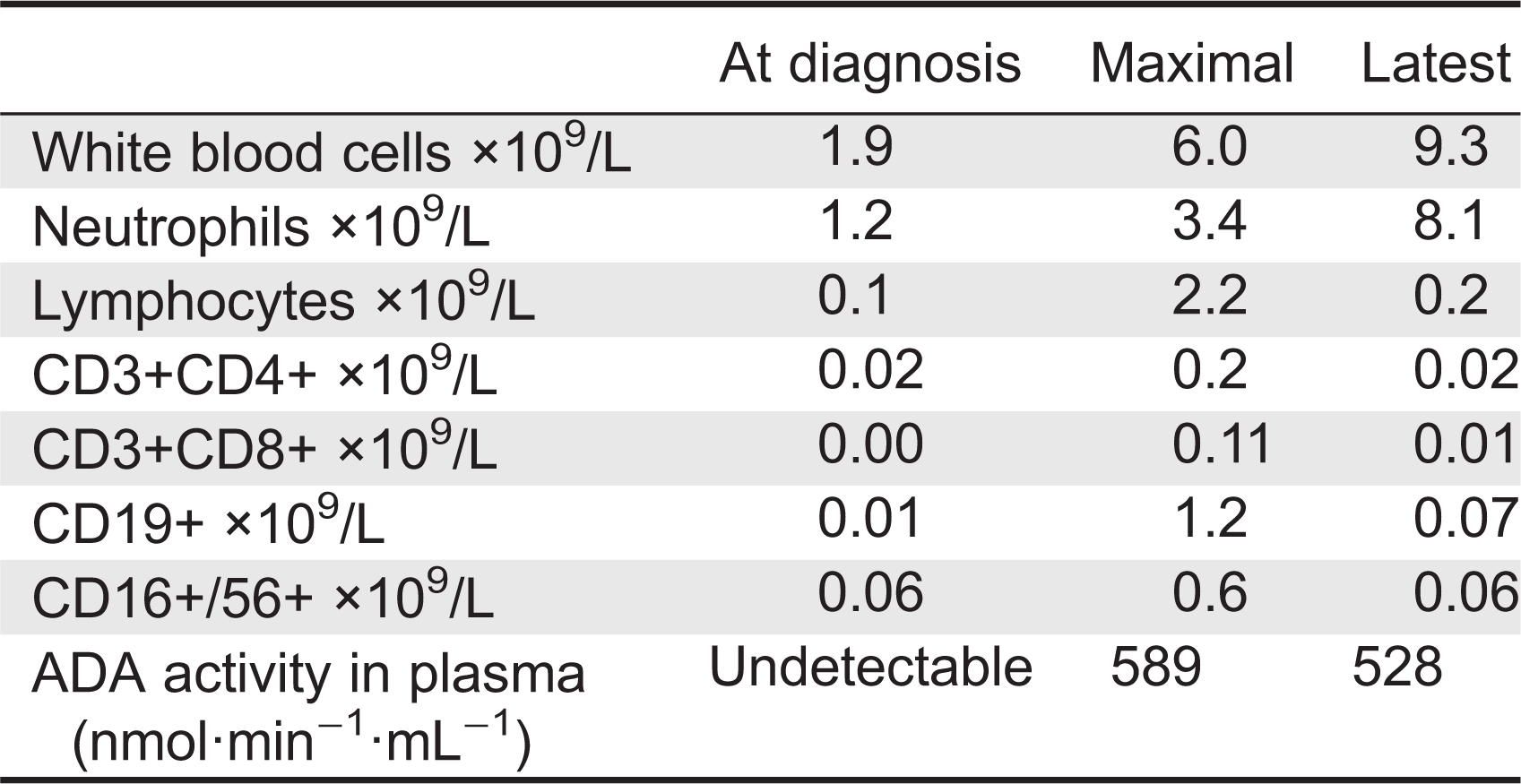
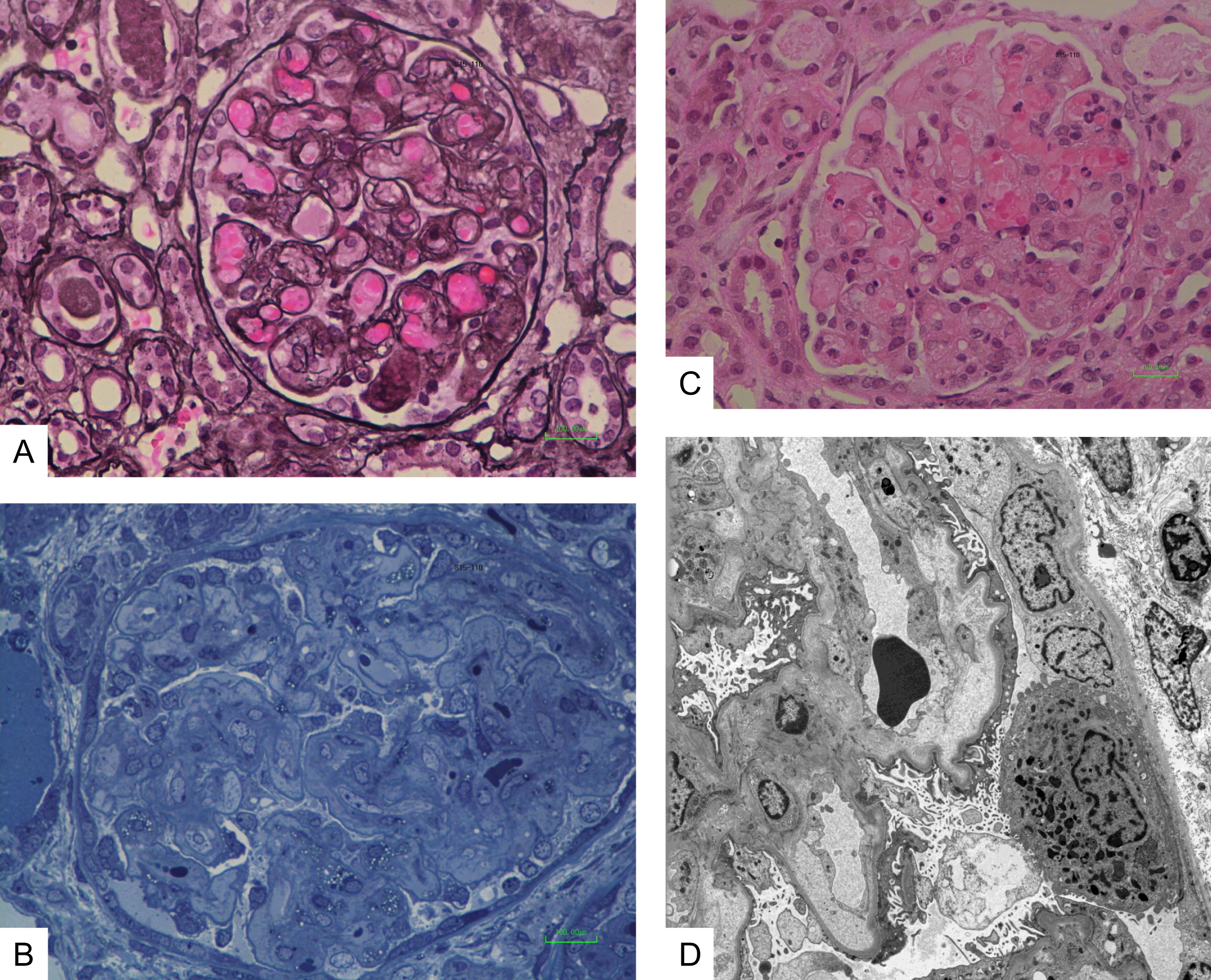
At 6 years of age the patient was admitted following a 2-week history of vomiting, fatigue, and increased bruising with normal urine output and no diarrhea. The patient had received IVIG 3 weeks earlier, was reported to have recovered from a mild upper respiratory tract infection, and had normal complete blood count, kidney functions, and urine. Upon admission, the patient was afebrile and normotensive. Initial laboratory investigations revealed markedly elevated creatinine 500 µmol/L (normal 9–55 µmol/L), urea 50 mmol/L (normal 3.0–7.0 mmol/L), and phosphate 3.08 mmol/L (normal 1.30–1.75 mmol/L) as well as low hemoglobin 49 g/L (normal 105–135 g/L) and ionized calcium 0.69 mmol/L (normal 1.10–1.30 mmol/L) with normal coagulation. Blood gas revealed the patient's pH was 7.49 with a base excess of 12.9 mmol/L. Urinalysis showed 2+ blood and protein. All microbiology studies were negative. There was an E. coli O157:H7 outbreak occurring in the patient's community 2 months prior to her presentation. However there was no known direct contact of the patient with affected individuals nor was Shiga-toxin producing E. coli isolated from the patient's stools at the time of her acute presentation. All viral cultures (nasopharyngeal and stool) as well as blood cultures remained negative. There was also no history of ingestion of ethylene glycol, methanol, drugs, or other toxins. The patient was treated briefly with ceftriaxone and piperacillin-tazobactam. After 3 days in hospital, she developed nonbloody diarrhea and thrombocytopenia (nadir of 84 × 109/L), which resolved after 2 days. A kidney biopsy revealed mesangiolysis and replication of the basement membrane with insudation of plasma into the mesangium and into the glomerular capillary wall (Figure 1A). There was also thickening of the loops with narrowing and occlusion of the loops by the replicated basement membrane, matrix, and cells (Figure 1B). Some loops were occluded by erythrocytes and platelet aggregates (Figure 1C). Electron microscopy confirmed the glomerular mesangiolysis and the accumulation of a pale matrix within the glomerular basement membrane (GBM) with elevation of the endothelial cells and replication of the GBM (Figure 1D). These features were consistent with a thrombotic microangiopathy. There was no evidence of tubulo-interstitial nephritis. The patient was treated by hemodialysis, with improved kidney function and normalization of the serum creatinine and urea. PEG-ADA and IVIG administration were not interrupted. C3 and C4 levels were within the normal range. ADAMTS13 (von Willebrand factor-cleaving protease) testing was negative. Mutations in genes coding for proteins involved in regulation or activation of the alternative complement activation pathway including factor H, I, membrane cofactor protein, and factor B or C3 were not identified. Antibodies to complement components or Factor H were not detected. Thus, the ADA-deficient patient described here has unequivocal clinical, laboratory, and histological characteristics of HUS with significant kidney disease.
Figure 1:
Discussion
Abnormal kidney function and histology, including accumulation of mesangial fibrillar matrix (mesangial sclerosis), obliterated glomeruli, and thickened capillary subendothelial layer were previously described in 6 ADA-deficient patients (Ratech et al. 1985). However, clinical details of the patients and whether they exhibited features of HUS were not provided (Ratech et al. 1985). Recently, 4 additional ADA-deficient patients with moderate to severe acute kidney failure, hemolytic anemia, and thrombocytopenia requiring dialysis or plasmapheresis were reported (Table 2). These patients were identified as suffering from atypical HUS, although detailed kidney histology was not provided (Nikolajeva et al. 2015). Hence, our patient is the first where the diagnosis of HUS is further supported by histological findings. Interestingly, the patient described here had early hypertension, albeit mild, several years before the development of HUS. Also, reminiscent of the findings in our patient, kidneys from ADA-deficient mice also display substantial red blood cell increase in the glomeruli and convoluted tubules (Blackburn et al. 1998) as well as decreased capillary lumen and mesangial hypercellularity (Dai et al. 2011) that could be prevented by correction of the metabolic abnormalities. Cumulatively, the data presented here suggest contribution of the ADA defect to the development of kidney damage and HUS.
Table 2:
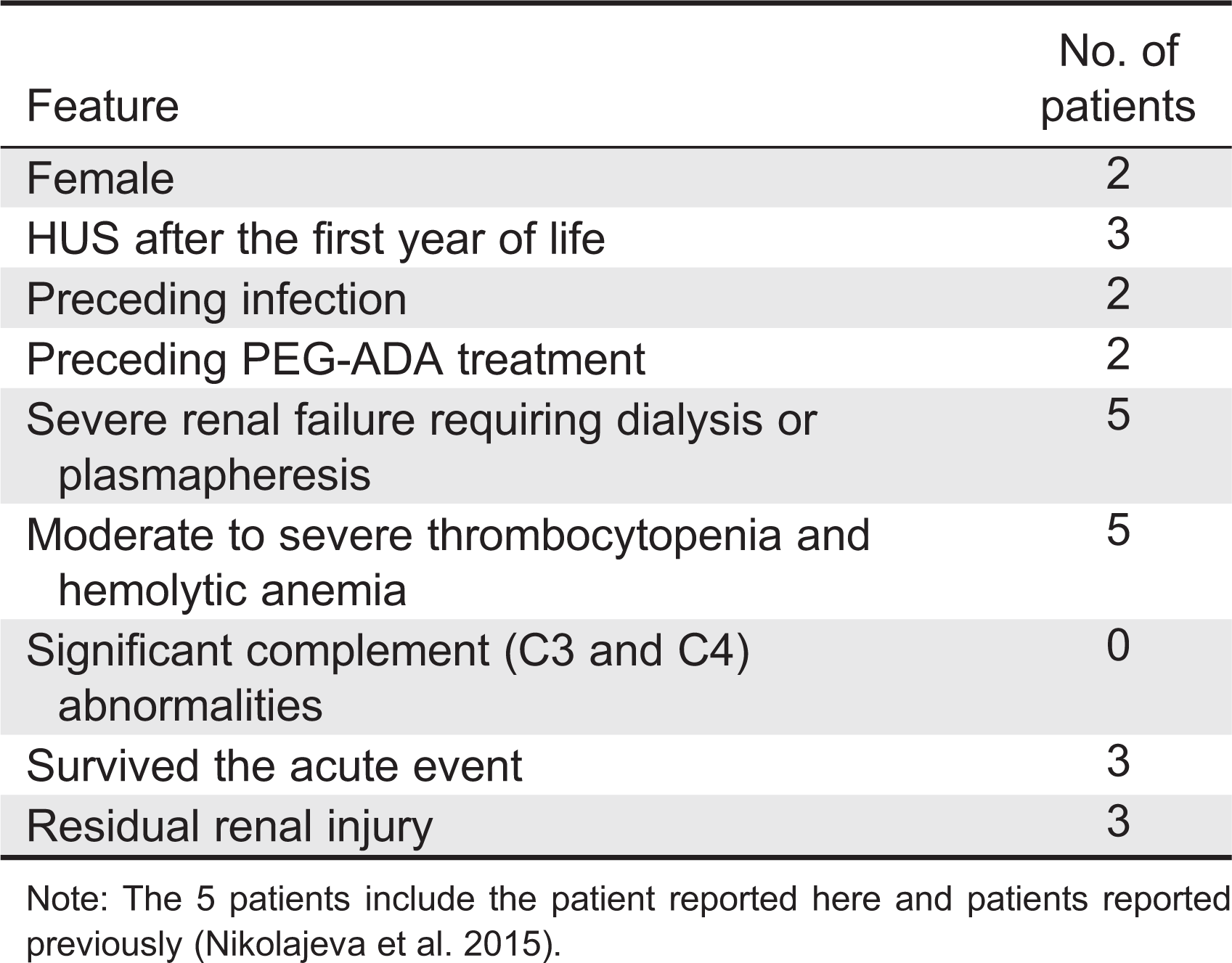
The pathogenesis of HUS in ADA-deficient patients is intriguing. It is unlikely that significant infections or autoimmunity were the culprit, as these conditions often do not resolve spontaneously in immune-compromised hosts. It is also unlikely that PEG-ADA caused kidney failure, even though it is a foreign protein, as only 1 of the 4 ADA-deficient patients previously reported with HUS had received PEG-ADA prior to the kidney damage (Table 2). Moreover, kidney function improved in our patient while she continued to receive PEG-ADA. Similarly, the ethnic diversity of the ADA-deficient patients suggests that the HUS was not caused by defects in other genes such as the complement gene family. Alternatively, it is possible that an intercurrent infection or post-infectious process reported in our patient and others exacerbated pre-existing kidney injury. We did not identify Shiga toxin E. coli O157 or evidence of pneumococcal infection in our patient. Regardless of the pathogenesis and somewhat reassuring, prompt intervention with dialysis reversed the kidney failure leading to improved kidney function.
In conclusion, our report demonstrates increased susceptibility of ADA-deficient patients for the development of HUS, expands the nonimmune abnormalities associated with ADA deficiency, and further emphasizes the vigilance required when caring for these patients.
Acknowledgements
EG is supported by the Donald and Audrey Campbell Chair for Immunology research.
REFERENCES
Blackburn M.R., Datta S.K., and Kellems R.E. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency J. Biol. Chem 1998 273 9 5093 -5100
Bollinger M.E., Arredondo-Vega F.X., Santisteban I., Schwarz K., Hershfield M.S., and Lederman H.M. Brief report: Hepatic dysfunction as a complication of adenosine deaminase deficiency N. Engl. J. Med 1996 334 21 1367 -1371
Brandt J., Wong C., Mihm S., Roberts J., Smith J., Brewer E., Thiagarajan R., and Warady B. Invasive pneumococcal disease and hemolytic uremic syndrome Pediatrics 2002 110 2 371 -376
Dai Y., Zhang W., Wen J., Zhang Y., Kellems R.E., and Xia Y. A2B adenosine receptor-mediated induction of IL-6 promotes CKD J. Am. Soc. Nephrol 2011 22 5 890 -901
Gaspar H.B., Aiuti A., Porta F., Candotti F., Hershfield M.S., and Notarangelo L.D. How I treat ADA deficiency Blood 2009 114 17 3524 -3532
Grunebaum E., Cohen A., and Roifman C.M. Recent advances in understanding and managing adenosine deaminase and purine nucleoside phosphorylase deficiencies Curr. Opin. Allergy Clin. Immunol 2013 13 6 630 -638
Grunebaum E., Cutz E., and Roifman C.M. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency J. Allergy Clin. Immunol 2012 129 6 1588 -1593
Husain M., Grunebaum E., Naqvi A., Atkinson A., Ngan B.Y., Aiuti A., and Roifman C.M. Burkitt's lymphoma in a patient with adenosine deaminase deficiency-severe combined immunodeficiency treated with polyethylene glycol-adenosine deaminase J. Pediatr 2007 151 1 93 -95
Keir L. and Coward R.J. Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy Pediatr. Nephrol 2011 26 4 523 -533
Nikolajeva O., Worth A., Hague R., Martinez-Alier N., Smart J., Adams S., Davies E.G., and Gaspar H.B. Adenosine deaminase deficient severe combined immunodeficiency presenting as atypical haemolytic uraemic syndrome J. Clin. Immunol 2015 35 4 366 -372
Ratech H., Greco M.A., Gallo G., Rimoin D.L., Kamino H., and Hirschhorn R. Pathologic findings in adenosine deaminase-deficient severe combined immunodeficiency. I. Kidney, adrenal, and chondro-osseous tissue alterations Am. J. Pathol 1985 120 1 157 -169
Sauer A.V., Brigida I., Carriglio N., and Aiuti A. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency Front Immunol 2012 3 265
Sokolic R., Maric I., Kesserwan C., Garabedian E., Hanson I.C., Dodds M., Buckley R., Issekutz A.C., Kamani N., Shaw K., Tan B., Bali P., Hershfield M.S., Kohn D.B., Wayne A.S., and Candotti F. Myeloid dysplasia and bone marrow hypocellularity in adenosine deaminase-deficient severe combined immune deficiency Blood 2011 118 10 2688 -2694
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 4 • December 2015
Pages: 195 - 199
History
Received: 2 June 2015
Accepted: 15 July 2015
Accepted manuscript online: 21 July 2015
Version of record online: 21 July 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Anne Pham-Huy, Vy Hong-Diep Kim, Elizabeth Nizalik, Gabrielle Weiler, Jennifer Vethamuthu, and Eyal Grunebaum. 2015. Atypical hemolytic-uremic syndrome in a patient with adenosine deaminase deficiency. LymphoSign Journal.
2(4): 195-199. https://doi.org/10.14785/lpsn-2015-0010
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
