RelB deficiency causes combined immunodeficiency
Abstract
Background: Combined immunodeficiency (CID) presents in infancy with severe microbial infections due to either the depletion or dysfunction of lymphocytes. Several mutated genes have been implicated in causing this condition. These encoded proteins are involved in gene recombination, signal transduction from receptors to transcription factors, or they are critical for lymphocyte development. There remain 20%–30% of patients with similar phenotypes but with no known genetic aberration. The objective of this study was to define the molecular basis of CID in a group of patients.
Genotyping was performed using linkage panel chips, and the results were analyzed for parametric linkage. Whole genome sequencing was also performed. In vitro mitogen stimulation, flow cytometry, real time PCR, Western blotting, and cytokine ELISA were used to assess immunological status and signal transduction pathways.
We identified a homozygous mutation in the gene for the NFκB transcription factor RelB in 3 patients who suffered repeated infection despite the presence of circulating T and B cells. This mutation introduces a premature stop, resulting in an ablation of RelB expression. Evaluation of patient immune systems revealed reduced response to mitogens and an inability to maintain an adequate antibody response to immunizations.
Lack of RelB expression results in a clinical presentation of CID.
Statement of novelty: We describe RelB deficiency for the first time.
Introduction
The nuclear factor kappa-B (NFkB) family of transcription factors consists of 5 members: RelA (p65), RelB, Rel (c-Rel), and 2 precursor proteins NFkB1 (p105) and NFkB2 (p100), which are processed to p50 and p52, respectively (Sun 2011; Millet et al. 2013). Two major pathways of NFkB activation are recognized: the classical, mediated by RelA (and c-Rel); and the less understood alternative, mediated by RelB. Infectious agents (through toll receptors), inflammatory cytokines, and lymphocyte antigen receptor activation all activate NFkB activity (Sun 2011; Millet et al. 2013).
All the NFkB proteins contain a Rel homology domain necessary for DNA binding. However, trans-activation and nuclear localization domains are separated between proteins, requiring the formation of dimers to obtain an active complex with nuclear targeting ability. Classical pathway activation is regulated by the binding of inhibitory IκB proteins to Rel-A/p105 complexes, sequestering them in the cytoplasm (Sun 2011; Millet et al. 2013). Phosphorylation of IκB proteins by IKK kinases initiates IκB degradation, freeing the NFkB complexes to then enter the nucleus. In contrast, RelB activity is suppressed by the binding of NFκB2 p100; alternative pathway activation requiring inducible proteolytic processing of p100 to its p52 form (initiated by NIK and IKKα) to permit RelB nuclear entry (Sun 2011; Millet et al. 2013).
Although RelA is activated by a large number of receptors (e.g., TNFR, TLR4, TCR), RelB activity is observed downstream from only a handful (BAFF-R, CD40, TWEAK, Lymphotoxin-β-R), despite its wide expression. Furthermore, although these receptors utilize the alternative pathway, simultaneous activation of the classical pathway is observed. In addition to its receptor activated functions, the presence of RelB is proposed to be necessary for the regulation of other family members (Oeckinghaus et al. 2011; Sun 2011; Millet et al. 2013). RelB knockout mice show that the protein plays an important role in the development of secondary lymphoid tissue (Burkly et al. 1995; Weih et al. 1995). Furthermore, most RelB −/− mice die young, owing to overwhelming multi-organ inflammatory processes stemming from aberrant T-cell cytokine secretion and (or) antigen recognition.
Human primary immune deficiencies have previously been shown to arise from mutations in NFκB Essential Modulator (NEMO, which is also known as inhibitor of nuclear factor kappa-B kinase subunit gamma (IKKγ)), IκBα, and Inhibitor of NFκ-B kinase subunit beta (IKKβ, which is also known as IKK2) (Doffinger et al. 2001; Courtois et al. 2003; Janssen et al. 2004; Orange et al. 2004; Lopez-Granados et al. 2008; Picard et al. 2011; Pannicke et al. 2013; Schimke et al. 2013). We show here that RelB deficiency in humans is associated with a combined immunodeficiency (CID).
Methods
Patient data were compiled prospectively and retrospectively from medical records and were entered into the Canadian Centre for Primary Immunodeficiency Registry and tissue bank, which was approved by the SickKids Research Ethics board (protocol no. 1000005598). This includes consent and assent from patients and parents for genetic analysis and the collection of tissue including thymus. Patients were treated primarily at The Hospital for Sick Children, Toronto, Ontario.
Linkage analysis
MERLIN (Abecasis et al. 2002) was used for parametric linkage analysis. Minor allele frequency was established based in HapMap CEU (North Americans and Northern and Western European descent) and TSI (Italians from Tuscany) samples. The parametric logarithm base 10 of odds (LOD) score was calculated for a 1 centiMorgan (cM) grid along the chromosomes at disease allele frequency of 1%.
Whole genome sequencing
The whole genome of 1 patient (patient 2) was sequenced using the Complete Genomics (Mountain View, California, USA) platform.
T- and B-cell proliferative responses
Lymphocyte proliferative responses to mitogens (including phytohemagglutinin (PHA) and anti-CD3 antibodies) and to a panel of recall antigens (including candida, tetanus, herpes zoster, and cytomegalovirus) were determined by thymidine incorporation at day 3 or day 6. All assays were performed in triplicate and were compared with simultaneously stimulated random normal controls.
Immunoglobulin and specific antibody determinations
Serum concentrations of immunoglobulins were measured by nephelometry. Serum IgE concentration was measured by radioimmunoassay with the IgE PRIST kit (Pharmacia Diagnostics, Quebec, Canada). Levels of serum antibodies to tetanus were measured by ELISA and polio antibody titers were determined by complement fixation.
Real-time polymerase chain reaction (RT-PCR)
RT-PCR was performed on an ABI 7500 real-time PCR system (Applied Biosystems) using 0.1µg of cDNA with the SYBR select Master Mix (Life Technologies). We used the following primers:
GAPDH-F: GAAACTGTGGCGTGATGGC,GAPDH-R: CACCACTGACACGTTGGCAG,RELB-F: CCCTACGAGGACCTGGAGAT, andRELB-R: GCAGGAAGTGGTCCAGGAT.
Western blotting
Whole-cell lysates were prepared in a RIPA buffer and analyzed by Western blotting. Anti-RelA, c-Rel, IκBα, NFκB1 p100, actin, Giα, C-terminal RelB (sc-226), and N-terminal RelB antibody (sc-38007) were purchased from Santa Cruz Biotechnology Inc. (Dallas, Texas, USA). Anti N-terminal RelB (04-1077) was also purchased from EMD Millipore (Billerica, Massachusetts, USA). Where necessary, expression levels were quantified by densitometry scanning and normalized by comparison with matching control protein levels. All blots were repeated at least twice.
Results
Case reports
Patient 1 was born after cesarean section at 41 weeks to consanguineous parents of Irish descent. At the age of 4 months, patient 1 began experiencing persistent cough and had 3 episodes of pneumonia, all of which required hospitalization. In addition, he suffered multiple episodes of otitis media and required tympanostomy tubes.
Patient 2, the younger brother of patient 1, was born at term by scheduled cesarean section. From the age of 1 month he had a chronic cough and repeated upper respiratory infections. At the age of 13 months, he was admitted for ecthyma gangrenosum which was confirmed by skin biopsy (Figure 1). He has suffered multiple episodes of lobar pneumonia that required antibiotic treatment and hospitalization, as well as failure to thrive. He was noticed to have intermittent swelling of his knees with limping.
Figure 1:
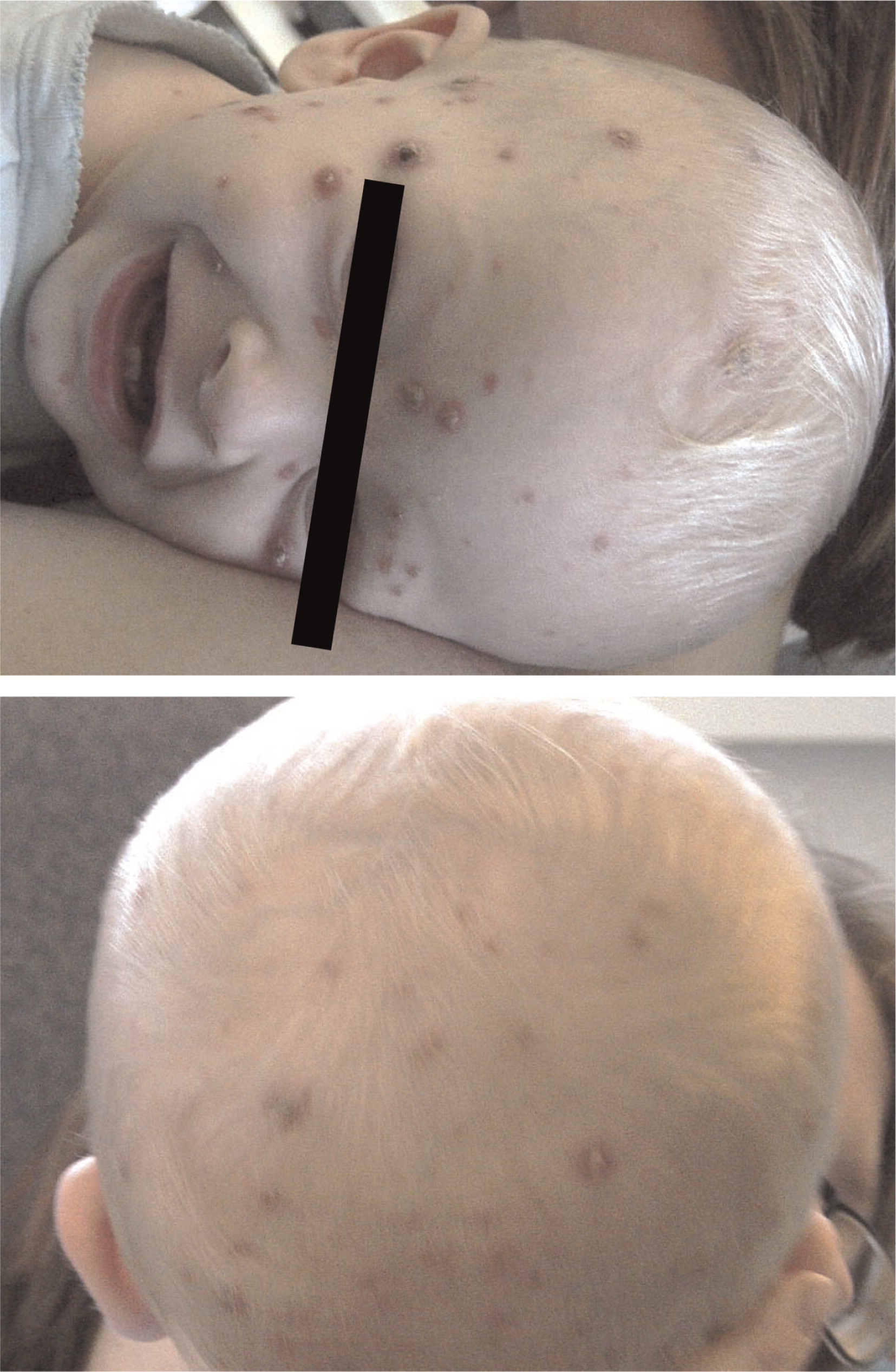
Patient 3, a first cousin of patients 1 and 2, was born at term. Immediately after birth he developed pneumothorax and was kept in hospital for 7 days. Growth and development have been slow, and he remains small at less than 10th percentile for height and weight. He has a chronic cough and has had reactive airway disease since infancy. He suffered multiple episodes of otitis media as well as pneumonia and 1 episode of urinary tract infection. He had visible tonsillar tissue as well as palpable lymph nodes.
Evaluation of the immune system
All patients displayed normal numbers of circulating white blood cells upon presentation. Immunophenotyping revealed that circulating lymphocyte numbers were normal to elevated in all 3 patients. (Table 1). Although the number of circulating CD3+ T cells was within normal range for age, CD4+ T cells were somewhat increased, resulting in aberrant CD4:CD8 ratios of 4–10:1 (controls being 1.8–2.5:1). CD20 positive B-cell numbers were also significantly elevated, whereas CD56 positive natural killer cells were normal.
Table 1:
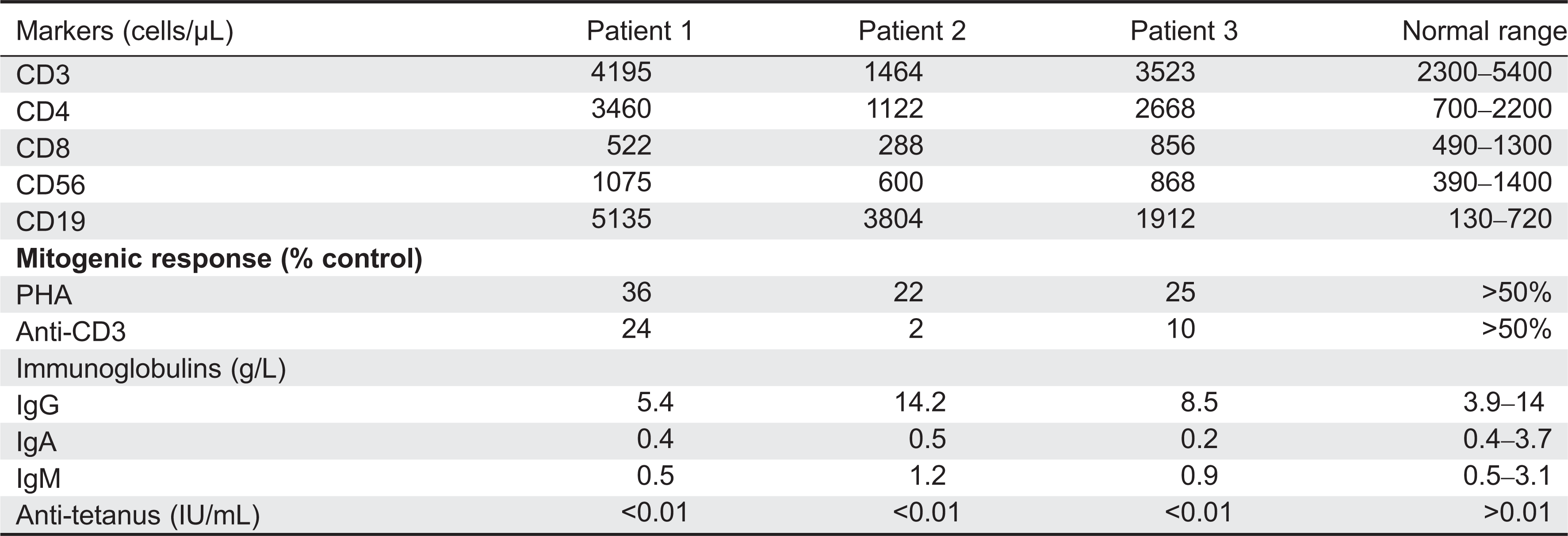
Despite the number of circulating T cells, in-vitro responses to PHA were less than 50% of normal controls and in-vitro responses to antigens including candida, zoster, simplex, and cytomegalovirus were completely flat (not shown). Total serum immunoglobulin levels were normal, but the ability to produce specific antibodies in vivo appeared to be significantly impaired (Table 1).
Linkage analysis
Four family members, corresponding to the 2 male affected siblings and the parents, were analyzed using Illumina linkage panel chip (5913 single nucleotide polymorphisms (SNPs)). All of the 4 samples had a call rate greater than 98.9%. The self-reported gender and pedigree structure were confirmed by genotype data analysis using PLINK (Purcell et al. 2007).
MERLIN (Abecasis et al. 2002) was used for parametric linkage analysis. The minor allele frequency of the 4135 common SNPs present in both HapMap III and the Illumina linkage panel were estimated based on HapMap CEU1 samples. Disease was assumed to be autosomal recessive with full penetrance as: 0.0, 0.1, and 1.0 (no mutated allele, 1 mutated allele, and 2 mutated alleles, respectively).
The parametric LOD score was calculated for a 1 cM grid along the chromosomes at disease allele frequency of 1%. Four chromosome regions were identified with a maximum LOD score larger than 2 (hg19 genomic coordinates) (Figure 2):
Chromosome 3 (i): start = rs11720298, 70.9405 cM, 51 581 509 (hg19); end = rs1472653, 80.094 cM, 59 585 592 (hg19);Chromosome 3 (ii): start = rs1317244, 120.54 cM, 112 053 565 (hg19); end = rs1052620, 144.177 cM, 136 574 521 (hg19)Chromosome 18: start = rs736839, 70.0197 cM, 46 528 065 (hg19); end = rs1539964, 97.8493 cM, 67 625,285 (hg19)Chromosome 19: start = rs1055099, 69.3765 cM, 43 979 589 (hg19); end = rs1017379, 91.09 cM, 52 457 306 (hg19)
Figure 2:
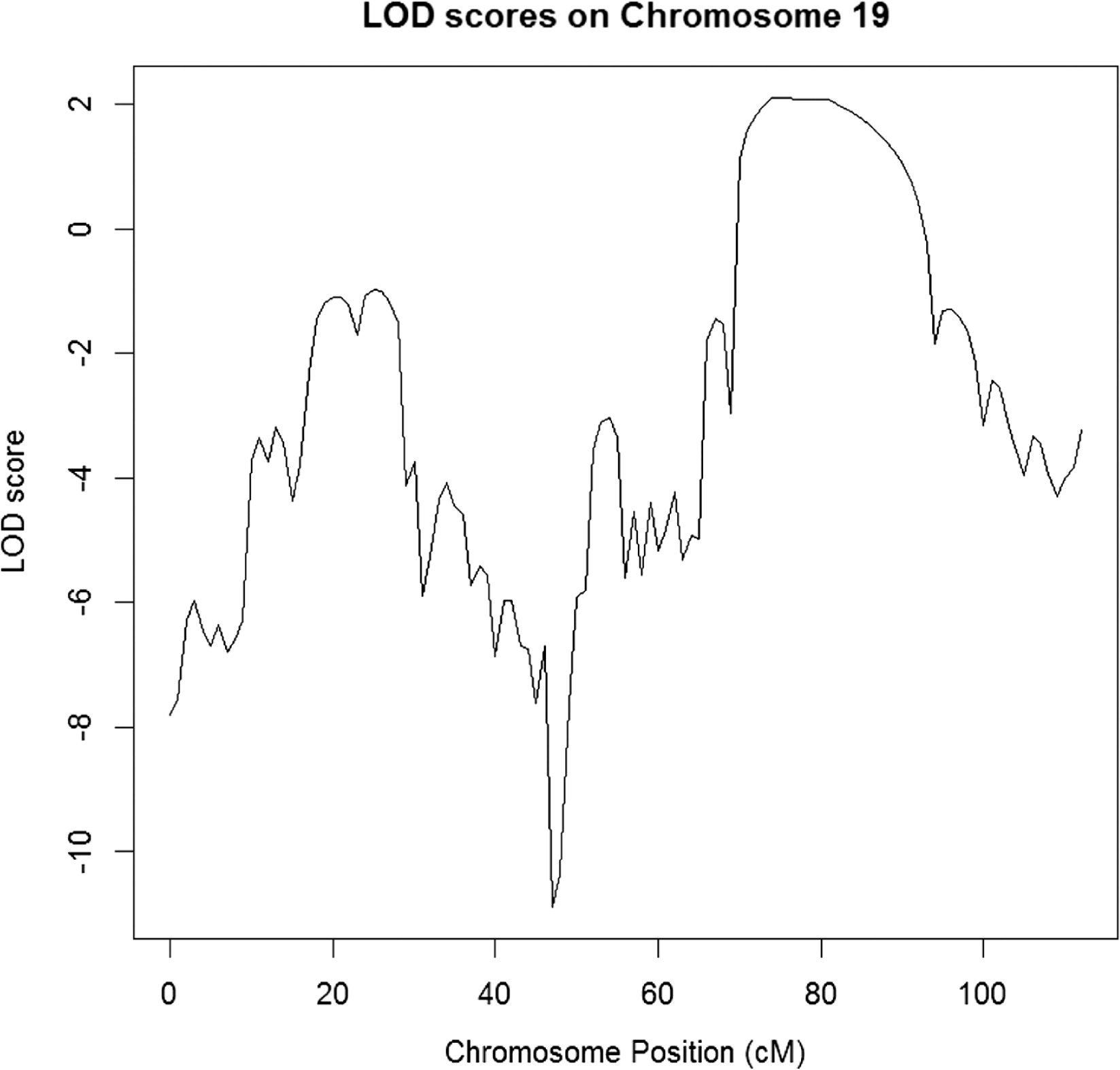
Whole genome sequencing
The whole genome of patient 2 was sequenced using the Complete Genomics (Mountain View, Calif.) platform. The concentration of genomic DNA sample was measured with picogreen, and approximately 10 mg of DNA was submitted to Complete Genomics for whole genome sequencing. Complete Genomics employs high-density DNA nanoarrays that are concatemers of mate pair reads, each approximately 500 bp long. Base identification was performed using a non-sequential, unchained read technology known as combinatorial probe-anchor ligation (cPAL). Each mate pair includes 35 nt of genomic DNA sequence as well as adaptor sequences required for cPAL sequencing; the average mate gap length is 300 bp.
The resulting whole genome sequence had a fully called genome fraction (i.e., excluding sites with no-calls) of 96.7% and a fully called exome fraction of 98.3%. The genome fraction at weighted coverage ≥10 was 97.7% and the corresponding exome fraction was 98.8%; in particular, >60% of the genome was covered at >40× – summary statistics indicative of a high-quality sequencing run.
Disease variant identification
The search for the disease variant(s) was initiated by applying the following filters on single nucleotide variants, small indels, and small block substitutions: (i) restrict to the 4 linkage peaks; (ii) restrict to high-quality variants; we selected variants with the varQuality field of the Complete Genomics MasterVarBeta file equal to VQHIGH (corresponding to VAF, i.e., variable allele fraction model, quality score for both alleles equal or greater than 20 db for homozygous calls, and equal or greater than 40 for other calls); and (iii) restrict to variants annotated as “coding” or “splicing” by either Complete Genomics’ annotations or in-house annotations based on Annovar (Wang et al. 2010).
This procedure resulted in 50–500 variants per peak. We further restricted to homozygous variants, assuming a recessive model, and excluded synonymous variants. Variants were manually reviewed, taking into account: (i) variant frequency in reference databases (1000 Genomes, NHLBI-ESP, Complete Genomics reference panel) (Abecasis et al. 2010; Tennessen et al. 2012) and presence in dbSNP137; (ii) residue-level conservation (PhyloP score) and local sequence conservation (multi-species alignments as provided by the UCSC browser); (iii) variant effect on coding sequence (e.g., missense, stop-gain, splicing); and (iv) gene function, provided by Gene Ontology (Ashburner et al. 2002), and associated phenotypes, provided by HPO/OMIM (OMIM descriptions as processed into a controlled vocabulary by the Human Phenotype Ontology) and MPO/MGI (Mammalian Phenotype Ontology annotations for mouse genes provided by Mouse Genome Informatics) (Smith et al. 2005; Robinson et al. 2008).
We identified a homozygous stop-gain in the gene RelB (chr19: 45,535,991). Both alleles had a high-quality score (variable allele fraction model: 111 and 981 db; equal allele fraction model: 50 and 52 db) and the variant was novel according to all reference databases (dbSNP, 1000 Genomes, NHLBI-ESP, Complete Genomics reference panel). Inspection of the locus in UCSC did not reveal any anomaly. In addition, NHLBI-ESP has not reported any stop-gain or splicing variant for RelB in over 5000 subjects, suggesting that the gene is heavily under negative selection against loss of function variants; it is specifically interesting that no loss of function altering the final exons were found either.
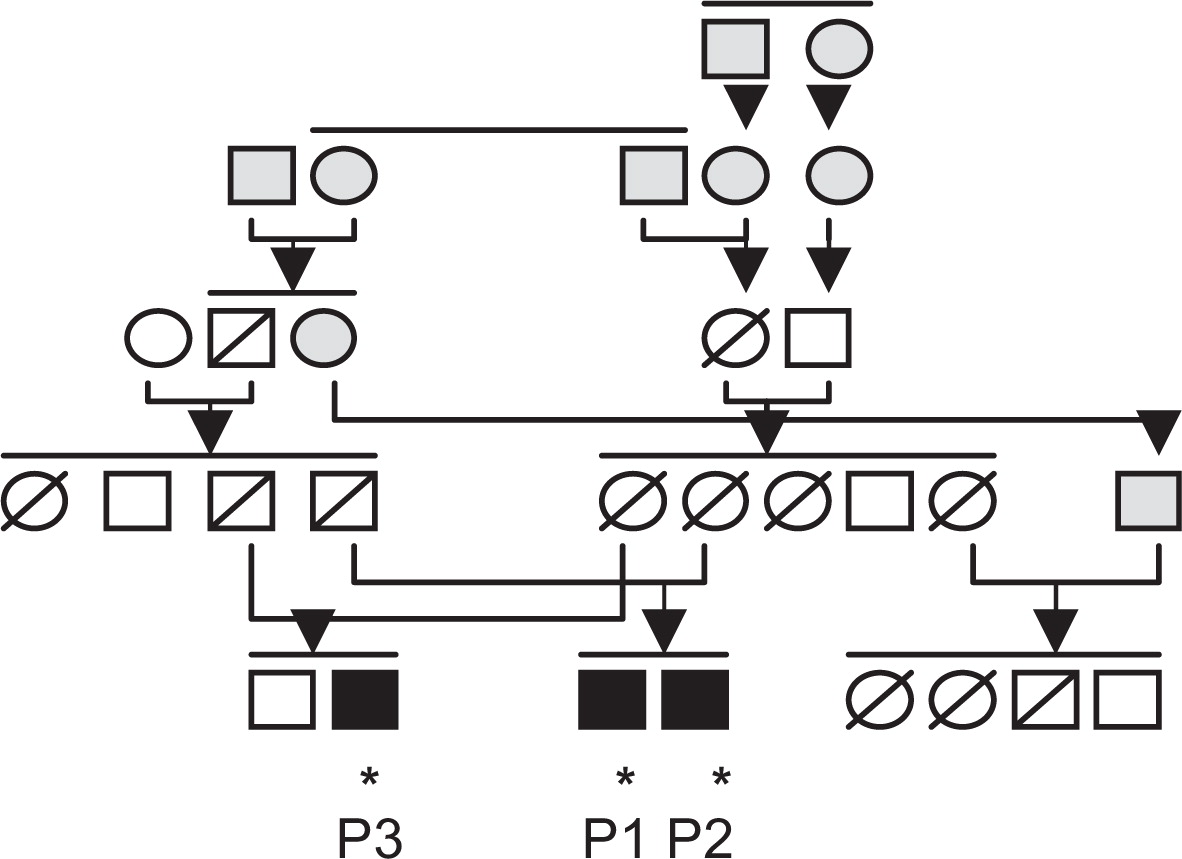
Subsequently, the stop-gain mutation was confirmed by Sanger sequencing and found to segregate with disease following the analysis of 21 family members (Figure 3). All 3 affected children were homozygous for the RelB mutation, all 4 parents were heterozygous, and unaffected siblings and close relatives were either heterozygous or carrying 2 wild-type alleles.
Figure 3:
Quantification of RelB mRNA and protein levels
Relative levels of RelB mRNA were assessed by RT-PCR analysis in 2 patients to determine whether the stop-gain mutation affected RelB mRNA transcription. Total RNA was isolated from Epstein–Barr virus (EBV) transformed B-cell lines (normal control, patient 2, and patient 3) and reverse transcription carried out with SuperScript III RT and Oligo(dT)12–18 primer. Primer pairs were checked for specificity by standard PCR. Relative quantification was performed using the standard curve method. The normal control EBV cDNA was used as the calibrator and GAPDH as an endogenous control. Results were determined as the mean and standard deviation for 5 replicate samples within a single experiment. In both patients examined, RelB mRNA levels were found to be lower than normal control (Table 2).
Table 2:

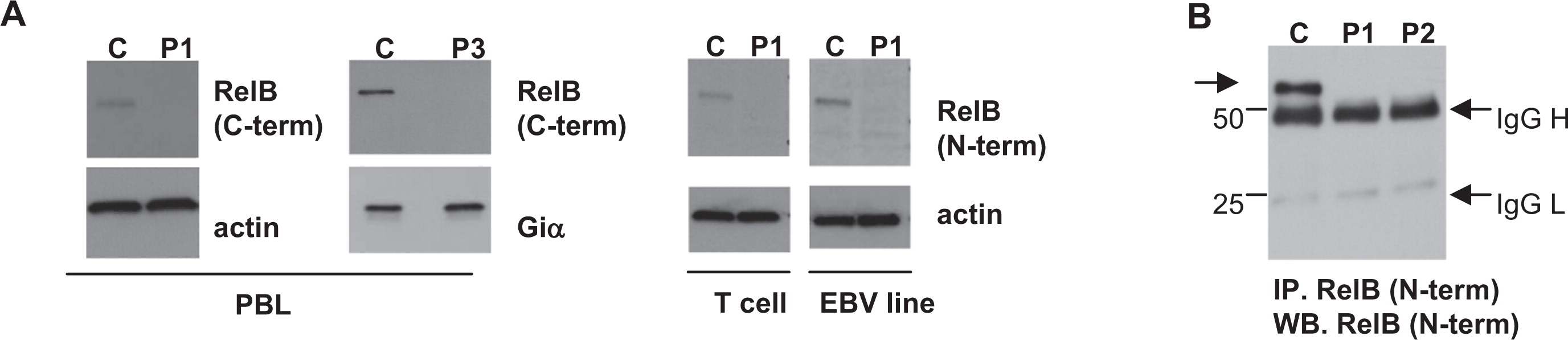
The mutation identified in RelB created a premature stop codon that blocked translation of the last 3 coding exons, shortening the protein by approximately one-third (terminating at amino acid 396) and potentially creating a protein devoid of transcription activating properties, owing to the deletion of its C-terminal nuclear localization sequence (Figure 4). Western blotting with antibodies against the RelB C-terminus, predicted to be deleted by the mutation, revealed no detectable RelB expression in the peripheral blood lymphocyte (PBL) of patient 1 and patient 3 (Figure 5). Furthermore, antibodies against the retained RelB N-terminal region also failed to detect any RelB protein in patient lysates. Enrichment by immunoprecipitation with anti-N terminus antibody and subsequent Western blotting similarly failed to detect any potential RelB fragment that would be coded by amino acids 1–396 if translated (predicted fragment, 43 kDa; wild type, 62 kDa) (Figure 5B).
Figure 4:
Figure 5:
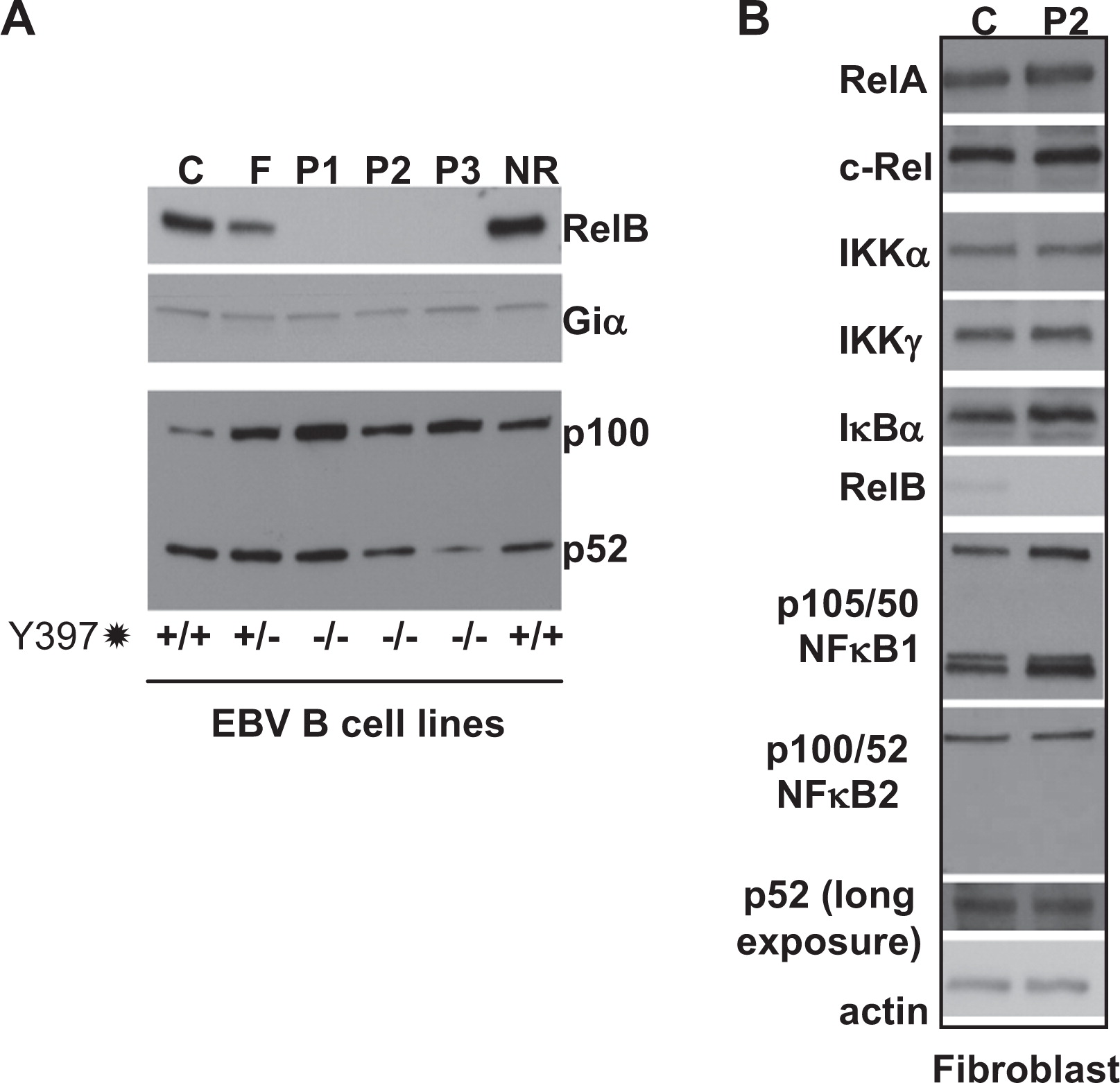
In contrast to RelB, all other components of the NFκB alternative and classical signaling pathways that appeared to be normally expressed were examined in patient EBV B cells and primary fibroblasts (Figure 6). Thus, the premature stop codon appears to effectively ablate RelB expression in these patients, resulting in a “knockout” phenotype.
Figure 6:
Discussion
We describe here a novel type of CID associated with a mutation in the RelB gene. Patients presented with repeated infections and failure to thrive, hallmarks of severe combined immunodeficiency (SCID). As these patients have normal to increased (yet dysfunctional) numbers of circulating T cells, they would be classified as leaky SCIDS or a CID. Occasionally, these types of patients may have autoimmune manifestations such as Omenn syndrome or colitis, yet management and outcome are similar to SCID in most cases (Roifman et al. 2012).
Patient consanguinity facilitated linkage analysis, several clear peaks with significant LOD scores were identified that ultimately helped pinpoint a homozygous mutations in RelB on chromosome 19. This premature stop codon mutation leads to decreased levels of RelB mRNA, presumably due to message instability. Furthermore, the RelB protein was undetectable in patient lymphocytes, suggesting that any potential fragment that may be translated is also unstable and subsequently degraded.
RelB −/− knockout mice die young of overwhelming inflammation in multiple organs owing to a T-cell dependent infiltration of tissues by myeloid lineage cells (although the exact mechanism is unclear) (Weih et al. 1995, 1996). In contrast, humans with RelB deficiency do not develop this T-cell driven disease, but rather suffer repeated infections and persist with chronic lung and gastro-intestinal symptoms. Patients with defects of NEMO or IκBα in the related NFκB classical pathway may present with anhidrotic ectodermal dysplasia; however, this was not seen in our patients. RelB deficient patients did not display atypical mycobacterial susceptibility either, as observed in classical pathway deficiencies.
Invariably, all 3 patients had failure to thrive, a typical manifestation of combined immunodeficiency. Evaluation of the immune system revealed dysfunctional T cells that were unresponsive to mitogens or antigens in-vitro, and humoral immunity was also severely affected, with a failure to respond to childhood vaccines. Ultimately, symptoms were severe enough to warrant treatment with hematopoietic stem cell therapy.
The fact that RelB, a member of the alternative pathway of NFκB activation, appears so critical for the function of the immune system is surprising. RelB activity has been reported in signaling pathways downstream from only a few receptors: BAFF-R, CD40, TWEAK, and Lymphotoxin-β-R, suggesting that its deletion might have caused minimal phenotype. It is likely that the immune defects apparent in RelB-deficient humans are due to a wider effect of RelB on the classical NFκB pathway (involving RelA and c-Rel) through cross-regulation of activation and expression in addition to the function of RelB within the alternate pathway.
ACKNOWLEDGEMENTS
This work was supported by the Program for Immunogenomics and the Canadian Centre for Primary Immunodeficiency, the Jeffrey Modell Foundation, and Immunodeficiency Canada. The Centre for Applied Genomics at SickKids was supported by Genome Canada through the Ontario Genomics Institute, Canada Foundation for Innovation, and the Ontario Ministry of Research and Innovation.
Footnote
1
CEU are Utah residents with Northern and Western European ancestry from the CEPH (Centre d'Etude du Polymorphisme Humain), which is the organization that collected the information.
REFERENCES
Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., and McVean G.A. A map of human genome variation from population-scale sequencing. 1000 Genomes Project Consortium Nature. 2010 467 7319 1061 -1073
Abecasis G.R., Cherny S.S., Cookson W.O., and Cardon L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees Nat. Genet. 2002 30 1 97 -101
Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., Harris M.A., Hill D.P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J.C., Richardson J.E., Ringwald M., Rubin G.M., and Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat. Genet. 2002 25 1 25 -29
Burkly L., Hession C., Ogata L., Reilly C., Marconi L.A., Olson D., Tizard R., Cate R., and Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells Nature. 1995 373 6514 531 -536
Courtois G., Smahi A., Reichenbach J., Doffinger R., Cancrini C., Bonnet M., Puel A., Chable-Bessia C., Yamaoka S., Feinberg J., Dupuis-Girod S., Bodemer C., Livadiotti S., Novelli F., Rossi P., Fischer A., Israel A., Munnich A., Le Deist F., and Casanova J.L. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency J. Clin. Invest. 2003 112 7 1108 -1115
Doffinger R., Smahi A., Bessia C., Geissmann F., Feinberg J., Durandy A., Bodemer C., Kenwrick S., Dupuis-Girod S., Blanche S., Wood P., Rabia S.H., Headon D.J., Overbeek P.A., Le Deist F., Holland S.M., Belani K., Kumararatne D.S., Fischer A., Shapiro R., Conley M.E., Reimund E., Kalhoff H., Abinun M., Munnich A., Israel A., Courtois G., and Casanova J.L. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling Nat. Genet. 2001 27 3 277 -285
Janssen R., van Wengen A., Hoeve M.A., ten Dam M., van der Burg M., van Dongen J., van de Vosse E., van Tol M., Bredius R., Ottenhoff T.H., Weemaes C., van Dissel J.T., and Lankester A. The same IkappaBalpha mutation in two related individuals leads to completely different clinical syndromes J. Exp. Med. 2004 200 5 559 -568
Lopez-Granados E., Keenan J.E., Kinney M.C., Leo H., Jain N., Ma C.A., Quinones R., Gelfand E.W., and Jain A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency Hum. Mutat. 2008 29 6 861 -868
Millet P., McCall C., and Yoza B. RelB: an outlier in leukocyte biology J. Leukoc. Biol. 2013 94 5 941 -951
Oeckinghaus A., Hayden M.S., and Ghosh S. Crosstalk in NF-kappaB signaling pathways Nat. Immunol. 2011 12 8 695 -708
Orange J.S., Jain A., Ballas Z.K., Schneider L.C., Geha R.S., and Bonilla F.A. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation J. Allergy. Clin. Immunol. 2004 113 4 725 -733
Pannicke U., Baumann B., Fuchs S., Henneke P., Rensing-Ehl A., Rizzi M., Janda A., Hese K., Schlesier M., Holzmann K., Borte S., Laux C., Rump E.M., Rosenberg A., Zelinski T., Schrezenmeier H., Wirth T., Ehl S., Schroeder M.L., and Schwarz K. Deficiency of innate and acquired immunity caused by an IKBKB mutation N. Engl. J. Med. 2013 369 26 2504 -2514
Picard C., Casanova J.L., and Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency Clin. Microbiol. Rev. 2011 24 3 490 -497
Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., and Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses Am. J. Hum. Genet. 2007 81 3 559 -575
Robinson P.N., Kohler S., Bauer S., Seelow D., Horn D., and Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease Am. J. Hum. Genet. 2008 83 5 610 -615
Roifman C.M., Somech R., Kavadas F., Pires L., Nahum A., Dalal I., and Grunebaum E. Defining combined immunodeficiency J. Allergy. Clin. Immunol. 2012 130 1 177 -183
Schimke L.F., Rieber N., Rylaarsdam S., Cabral-Marques O., Hubbard N., Puel A., Kallmann L., Sombke S.A., Notheis G., Schwarz H.P., Kammer B., Hokfelt T., Repp R., Picard C., Casanova J.L., Belohradsky B.H., Albert M.H., Ochs H.D., Renner E.D., and Torgerson T.R. A novel gain-of-function IKBA mutation underlies ectodermal dysplasia with immunodeficiency and polyendocrinopathy J. Clin. Immunol. 2013 33 6 1088 -1099
Smith C.L., Goldsmith C.A., and Eppig J.T. The mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information Genome. Biol. 2005 6 1 R7
Sun S.C. Non-canonical NF-kappaB signaling pathway Cell. Res. 2011 21 1 71 -85
Tennessen J.A., Bigham A.W., O'Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Kang H.M., Jordan D., Leal S.M., Gabriel S., Rieder M.J., Abecasis G., Altshuler D., Nickerson D.A., Boerwinkle E., Sunyaev S., Bustamante C.D., Bamshad M.J., and Akey J.M. Evolution and functional impact of rare coding variation from deep sequencing of human exomes Science. 2012 337 6090 64 -69
Wang K., Li M., and Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data Nucleic. Acids. Res. 2010 38 16 e164
Weih F., Carrasco D., Durham S.K., Barton D.S., Rizzo C.A., Ryseck R.P., Lira S.A., and Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family Cell. 1995 80 2 331 -340
Weih F., Durham S.K., Barton D.S., Sha W.C., Baltimore D., and Bravo R. Both multiorgan inflammation and myeloid hyperplasia in RelB-deficient mice are T cell dependent J. Immunol. 1996 157 9 3974 -3979
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 3 • September 2015
Pages: 147 - 155
History
Received: 27 February 2015
Accepted: 11 March 2015
Accepted manuscript online: 11 March 2015
Version of record online: 11 March 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Daniele Merico, Nigel Sharfe, Pingzhao Hu, Jo-Anne Herbrick, and Chaim M. Roifman. 2015. RelB deficiency causes combined immunodeficiency. LymphoSign Journal.
2(3): 147-155. https://doi.org/10.14785/lpsn-2015-0005
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Establishing reference ranges for lymphocyte proliferation responses to phytohemagglutinin in patients with T cell dysfunction
2. Whole exome and genome sequencing for Mendelian immune disorders: from molecular diagnostics to new disease variant and gene discovery
3. Heterozygous mutations in RelB can be associated with immune dysregulation and lymphoma
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
