The history and evolution of immunoglobulin products and their clinical indications
Abstract
The history of providing antibodies to treat diseases began in the 19th century with the discovery of tetanus and diphtheria toxins and the demonstration that immunity to tetanus and diphtheria infections could be transferred by immune sera. Characterization of the mediators of this immunity resulted in the discovery that antibodies are proteins that can be isolated and used to protect against infectious diseases. Development of a method to isolate antibodies from human plasma that could be safely injected into people initiated the development of human gamma globulin preparations to provide antibodies to patients with inherited antibody deficiencies. To overcome the limitations imposed by intramuscular injection of gamma globulin, intravenous gamma globulin preparations were developed that began to be used in a wide variety of clinical conditions. Thus the original clinical indication for infection prevention was expanded to several other indications that employ large doses to suppress inflammatory and autoimmune disorders. The most recent development in immunoglobulin therapy is the production of concentrated immune globulins for subcutaneous injection. Home infusions of subcutaneous immunoglobulin are increasingly used to treat immunodeficient patients and are being studied for other clinical applications.
Background
In 1859 Louis Pasteur proposed that infectious diseases were caused by germs (Ullmann 2007). In 1876, Robert Koch proved Pasteur’s germ theory by isolating the anthrax bacillus and later the turbercle bacillus (Encyclopedia Britannica 2014). Koch also set up a bacteriology laboratory that attracted scientists from around the world and resulted in the development of new methods to isolate and study pathogenic bacteria.
Koch formulated 4 basic criteria to prove that a disease was caused by a specific microorganism (Encyclopedia Britannica 2014):
1.
“A specific microorganism is always associated with a given disease.”
2.
“The microorganism can be isolated from the diseased animal and grown in pure culture in the laboratory.”
3.
“The cultured microbe will cause disease when transferred to a healthy animal.”
4.
“The same type of microorganism can be isolated from the newly infected animal.”
Koch and his collaborators contributed directly to the development of antibody therapy. While studying in Koch’s laboratory, the Japanese scientist Shibasaburo Kitasato produced the first pure culture of the anaerobic bacterium Clostridium tetani (Kitasato 1889) and discovered the existence of tetanus toxin in the filtrates of C. tetani cultures (von Behring and Kitasato 1890). These studies were published in the landmark paper entitled “On the mechanism of immunity to diphtheria and tetanus in animals” (von Behring and Kitasato 1890). In this article, the authors described the following experiment: (i) rabbits were vaccinated with a culture of virulent tetanus bacteria; (ii) blood was collected from the immunized rabbits; and (iii) 0.2–0.5 mL of the immune plasma injected into the abdominal cavity of mice protected them against lethal doses of C. tetani.
Kitasato and von Behring summarized their observations as follows:
“The blood of rabbits immune to tetanus has the ability to neutralize or destroy the tetanus toxin…This property exists also in extravascular blood and in cell-free serum…This property is so stable that it remains effective even in the body of other animals, so that it is possible, through blood or serum transfusions, to achieve an outstanding therapeutic effect…The property which destroys tetanus toxin does not exist in the blood of animals which are not immune to tetanus, and when one incorporates tetanus toxin into nonimmune animals, the toxin can still be demonstrated in the blood and other body fluids of the animal, even after its death.”
One week after the tetanus data were published, von Behring (1890) reported similar results with diphtheria bacteria in a paper entitled “Studies on the mechanism of immunity to diphtheria in animals”. von Behring and Kitasato were the first to discover antibody molecules in the serum of immunized animals and to demonstrate that these antibodies could neutralize diphtheria and tetanus toxins (Sri Kantha 1991). They also demonstrated that the antibodies were specific; tetanus antitoxin did not neutralize diphtheria toxin and vice versa (Sri Kantha 1991).
Antibodies are gamma globulins
Several investigators reported that antibodies were transmitted from mother to infant through the placenta. Investigators also observed that the concentration of diphtheria and tetanus antitoxins in fetal blood were the same as in maternal blood (McKhann and Chu 1933). Based on these observations, McKahnn and Chu (1933) studied the distribution of antibodies in the protein fractions of placental extracts. Extracts of placentas were fractionated by ammonium sulfate precipitation and tested for diphtheria anti-toxin. The globulin fractions contained diphtheria toxin-neutralizing activity (McKhann and Chu 1933). McKahnn and Chu (1933) also reported that placental extracts given by intramuscular injection early after exposure to measles “either completely protects children against measles or produces a modification of the disease”.
Further characterization of antibody molecules was provided by physical chemists in Sweden. Heidelberger and Pederson (1937) separated antibody proteins on the basis of their molecular weights by sedimentation in an ultracentrifuge. Using material provided by sedimentation, Tiselius (1937) further separated the globulins by their electrical charge using electrophoresis. Antibodies were found in the most slowly migrating globulin fraction, which was designated the “gamma globulin” fraction (Tiselius 1937). In further studies Tiselius and Kabat (1939) published an electrophoretic profile of the serum proteins as illustrated in Fig. 1.
Figure 1:
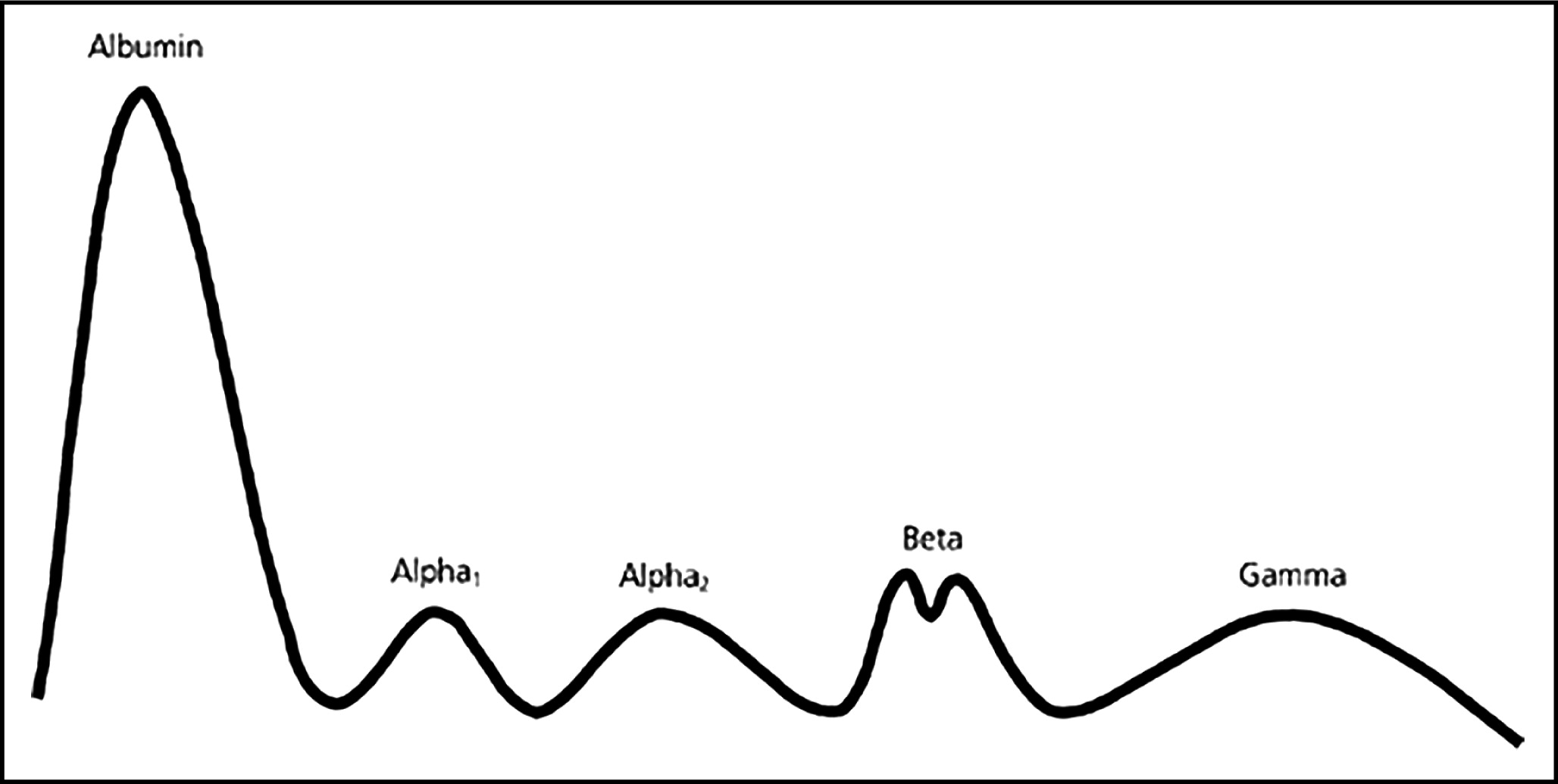
Immune serum globulin (an antibody concentrate)
The purification of serum globulins from human plasma by Cohn et al. (1944) provided material that could be safely injected into patients. Cohn et al. (1944) developed a series of cold ethanol precipitation steps that fractionated human plasma into classes of proteins, including the gamma globulin fraction commonly known as Cohn fraction II.
Enders analyzed fraction II for antibody activities and showed that 5 antibodies, including diphtheria antitoxin and mumps antibody, were concentrated 15–30 times compared with their concentration in the starting plasma (Enders 1944).
Stokes et al. (1944) demonstrated that immune serum globulin (ISG), formulated from Cohn fraction II at 165 mg/mL protein, protected or attenuated infections in children exposed to measles during epidemics in the winter of 1942 and the spring of 1943. They observed that almost all children 5 years of age and under were protected by doses of 2.0–2.5 mL of ISG injected intramuscularly, whereas complete protection of children 6–12 years of age required 4–5 mL of ISG. Stokes et al. (1944) also observed that some patients who had already developed symptoms of disease benefited from higher doses of ISG, and speculated that high doses delivered intravenously might provide therapeutic value in the early stages of measles infection.
In 1952, Bruton treated a child with undetectable serum gamma globulin levels who suffered from recurring pneumococcal infections. Subcutaneous infusions of 3.2 g/month ISG produced measureable circulating gamma globulin levels and completely eliminated the pneumococcal infections. These observations were confirmed by investigators in the United States who reported on 9 patients (Janeway et al. 1953) and in a report of 176 patients in the United Kingdom (Medical Research Council 1969).
Shown to be effective for post-exposure prophylaxis of measles and hepatitis A infections, ISG became the standard of care for patients with primary antibody deficiencies (Medical Research Council 1969; Schiff 1994). The standard dose was approximately 100–150 mg/kg. When children with measles infections were given ISG intravenously, they suffered from severe adverse reactions including fever, convulsions, restlessness, chills, and vasomotor collapse (Schiff 1994). Therefore, ISG administration was restricted to intramuscular or subcutaneous injection.
Intravenous immunoglobulin (IVIG)
The desire to deliver larger doses of ISG led to a series of manufacturing changes intended to produce an intravenously injectable immunoglobulin. Early IVIG preparations were produced from ISG by digestion with proteolytic enzymes to eliminate anticomplement activity. Anticomplement activity was believed to be the primary cause of adverse side-reactions when ISG was injected intravenously (Barandun et al. 1962). The first commercial IVIG was produced by digestion of ISG with pepsin. The principal component of pepsin-digested IVIG was a fragment that contained 2 antibody binding sites and was named the F(ab′)2 fragment (Fig. 2) (Schultze and Schwick 1962).
Figure 2:
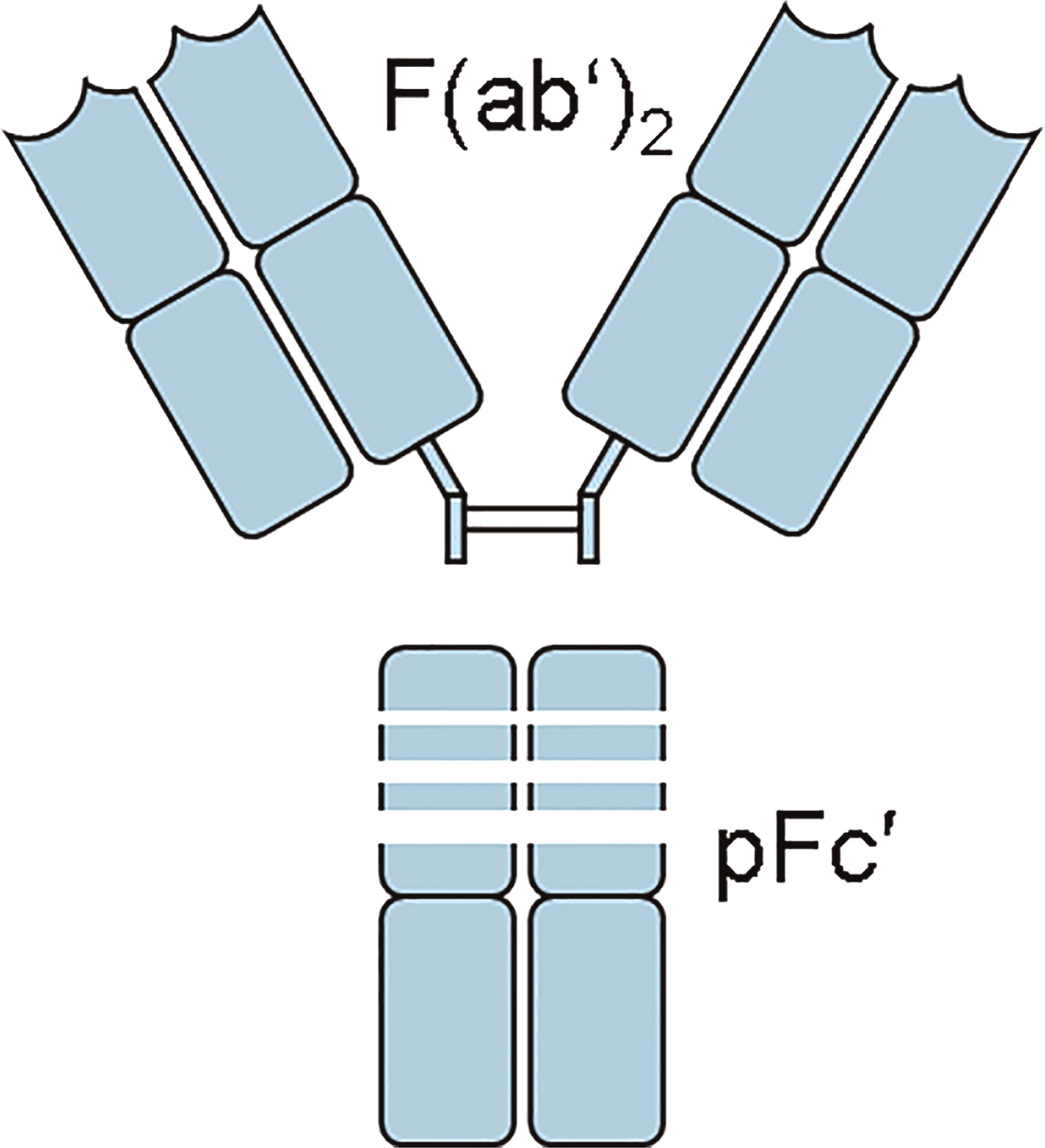
Other early IVIG manufacturing procedures involved treatment of ISG with enzymes or with chemical reagents that reacted with amino acids located in the complement binding site (Barandun et al. 1962; Sgouris 1967; Schroeder et al. 1980). Unfortunately, each of these modifications reduced important antibody activities (Pollack 1983; Kim et al. 1986; Bender and Hetherington 1987; Steele and Steele 1989) and shortened the half-lives of some antibodies (Janeway et al. 1968; Winston et al. 1982; Hagenbeek et al. 1987–1928). Today most commercial IVIGs are produced by first using Cohn cold ethanol fractionation followed by purification steps that remove IgA, IgM, and enzymes that cause side-effects.
In addition to manufacturing procedures, IVIG formulations have also changed over the years. Historically, most commercial IVIGs were formulated at pH 6.4–7.2 and 50 mg/mL. These products were usually freeze-dried in the presence of a stabilizer because IgG solutions at neutral pH are unstable and form precipitates during storage. The discovery that IgG solutions at low pH are physically stable has resulted in current IVIG products being formulated as liquids.
IVIG in the United States
The first IVIG licensed in the United States was produced from Cohn fraction II by reduction and alkylation (Schroeder et al. 1980). Using this product (Gamimune), Nolte et al. (1979) randomized 20 patients with primary immunodeficiency (PID) to receive IVIG or ISG. Doses were 150 mg/kg every 4 weeks of IVIG or 3.3 g/4 weeks of ISG. Since many subjects had chronic otitis media, sinusitis, and bronchitis, these acute infections, defined by “a spiking fever greater than 100.4 °F with or without chills, clinical evidence of a purulent infection, and positive cultures”, were monitored. The authors observed fewer acute infections during IVIG treatment but noted that the IVIG dose was significantly higher than the ISG dose (Nolte et al. 1979). A similar observation was made by Roifman et al. (1985).
The same product was studied by Amman et al. (1982) in a randomized 2-year crossover study. Thirty-four patients with serum IgG levels <300 mg/dL and who were unresponsive to immunization were studied. The patients were treated for 1 year with either IVIG or ISG at a dose of 100 mg/kg per month and then were treated for an additional year with the other product. No differences were observed in the incidence of acute infections but upper respiratory illnesses were more prevalent in IVIG recipients (85.3% vs 50.0% of patients, p < 0.006). This suggested that the dose rather than the rate of administration might be critical.
In a cross-over study of 12 patients with antibody deficiency and lung disease, Roifman et al. (1987) studied the impact of IVIG dose on clinical outcomes. Patients received either 600 mg/kg or 200 mg/kg of IVIG for 6 months and then were switched to the other dose for 6 months. The incidence of acute infections was substantially lower in patients receiving the higher dose when their serum IgG level reached ≥500 mg/dL. Lung function consistently improved in high dose recipients (Roifman et al. 1987).
In another crossover study, 12 PID patients received 500 mg/kg per month during the first year and 150 mg/kg per month during the second year, or vice versa. Fewer days of infection and less antibiotic treatment were required during high dose therapy. Compared with low-dose IVIG, high-dose IVIG reduced the number of fever days and improved lung function in some patients (Bernatowska et al. 1987).
A wide range of doses have been used in IVIG clinical trials of patients with PID. Most studies have shown a significant benefit when immunodeficient patients are treated with doses of 300 mg/kg per month or more (Schiff 1994).
IVIG in PID
All IVIGs licensed in the United States are studied in patients with PID. Early studies involved relatively few patients, several different doses, and different outcome parameters. In 1999, the US Food and Drug Administration (FDA) attempted to standardize IVIG clinical trials and proposed that new IVIGs be studied in a randomized, double-blind, non-inferiority study of 80 subjects with PID in which the safety and efficacy of the test product was to be compared with a US-licensed IVIG product over an observation period of 12 months (FDA Blood Products Advisory Committee Meeting, March 1999).
Subsequently, the FDA proposed that testing smaller numbers of subjects in an open-label, single-arm trial compared with a statistically modelled historical control would be sufficient to provide evidence of safety and efficacy (FDA Blood Products Advisory Committee Meeting, March 2000). In 2005, the FDA guidance document suggested that 40–50 subjects was an adequate number of patients, and safety and efficacy criteria were also defined (FDA/CBER Guidance for Industry 2005).
This guidance defined “infusional adverse events” (AE) as a safety parameter. Infusional AE are temporally associated with an infusion and occur during or within 1 h, 24 h, or 48 h following infusion of the test product. A principal safety endpoint was the observed proportion of infusions with 1 or more temporally associated AEs (including AEs that were determined not to be product related). The target for this endpoint was an upper one-sided 95% confidence limit of <0.40 (FDA/CBER Guidance for Industry 2005). The most recent FDA Guidance published in 2008 extended the time period for infusional AEs to 72 h and added instructions for conducting pharmacokinetic studies in pediatric patients (FDA/CBER Guidance for Industry 2008).
The principal efficacy parameter was defined as the rate of serious bacterial infections in adult and pediatric subjects over a period of 12 months (to avoid seasonal biases). The efficacy target was a statistical demonstration that the upper one-sided 99% confidence limit of serious bacterial infections is <1.0 per person per year. The FDA guidance documents defined the types of serious bacterial infections and the diagnostic criteria for each type.
The effect of these FDA guidance documents has been to standardize IVIG clinical trials in patients with PID in the United States. Table 1 summarizes results from recent IVIG clinical trials (Stein et al. 2009; Berger et al. 2010; Moy et al. 2010; Wasserman et al. 2012). The number of patients in each trial ranged from 46 to 80 and the mean dose per patient was 461–500 mg/kg. The same serious bacterial infection and adverse event endpoints were reported for each study.
Table 1:
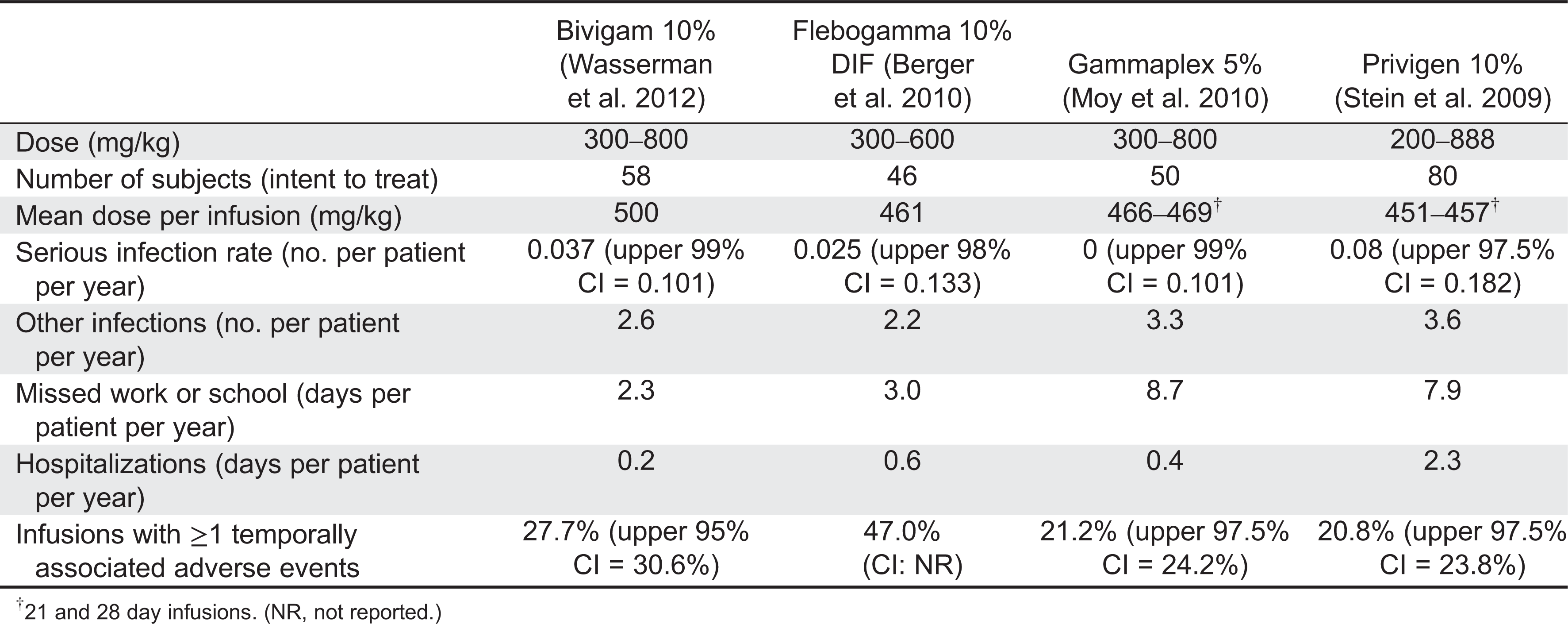
Immune thrombocytopenic purpura (ITP)
In 1981, Imbach et al. reported that the platelet count of a child with PID and thrombocytopenia increased within 24 h after treatment with 0.4 g/kg IVIG and continued to increase after 4 additional daily IVIG infusions (Imbach et al. 1981). These observations have been confirmed in controlled studies (Bussel et al. 1983) and the use of high-dose IVIG to treat acute ITP is well-established (Schiff 1994).
IVIG clinical applications
The reduction of the incidence and severity of acute infections patients with PID and the demonstration of immunomodulatory activity in ITP resulted in IVIG being used to treat a wide variety of clinical conditions. The use of high-dose IVIG treatment for ITP has been particularly influential in expanding IVIG clinical applications beyond prevention of infections. A partial list of clinical conditions treated with IVIG is shown in Table 2. IVIG treatment of the neurologic disorders, autoimmune disorders, and miscellaneous disorders listed in Table 2 all result from the ITP experience. However, most of the clinical uses listed in Table 2 have not been demonstrated to be efficacious in large clinical trials and are not approved indications. IVIG clinical applications that have been reviewed and approved by the FDA and the European Medicines Agency are listed in Table 3.
Table 2:
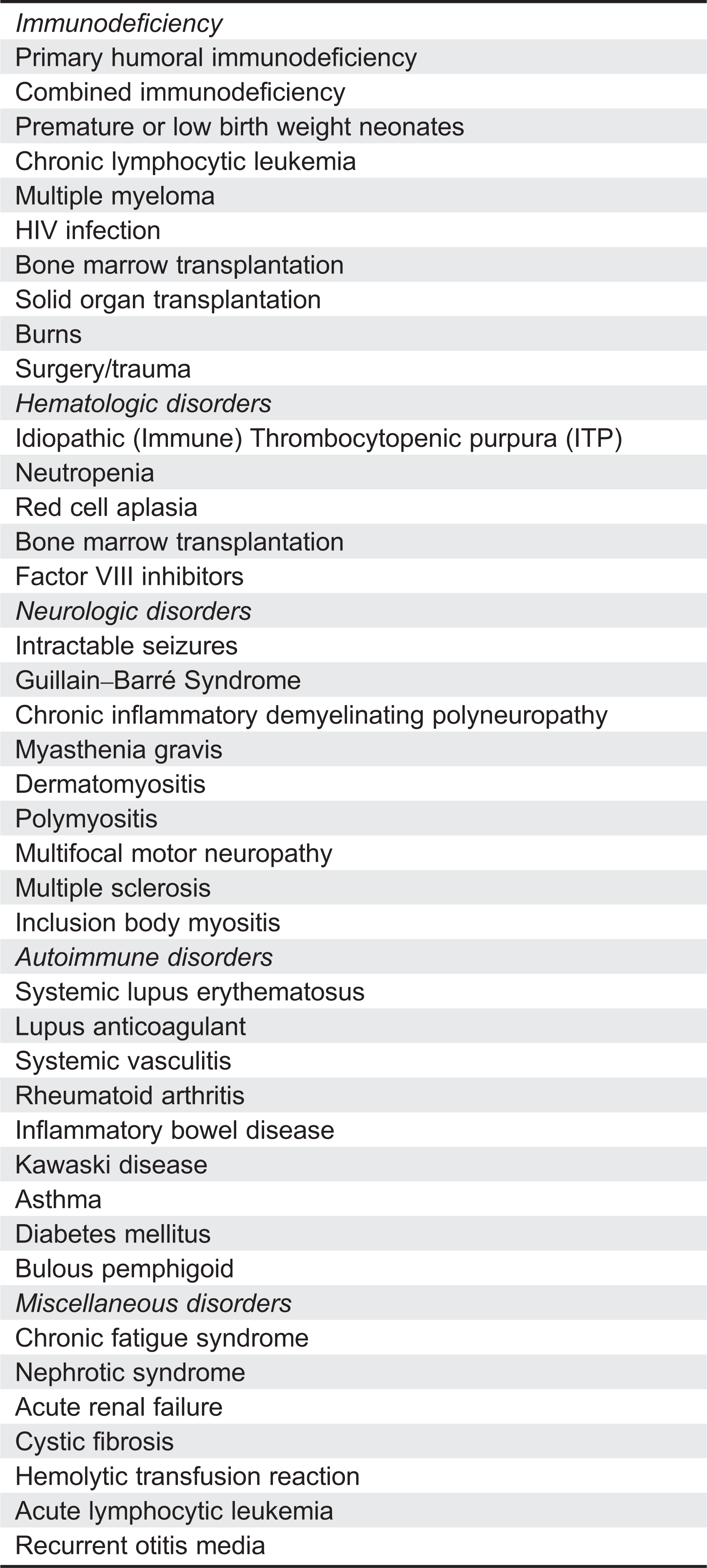
Table 3:
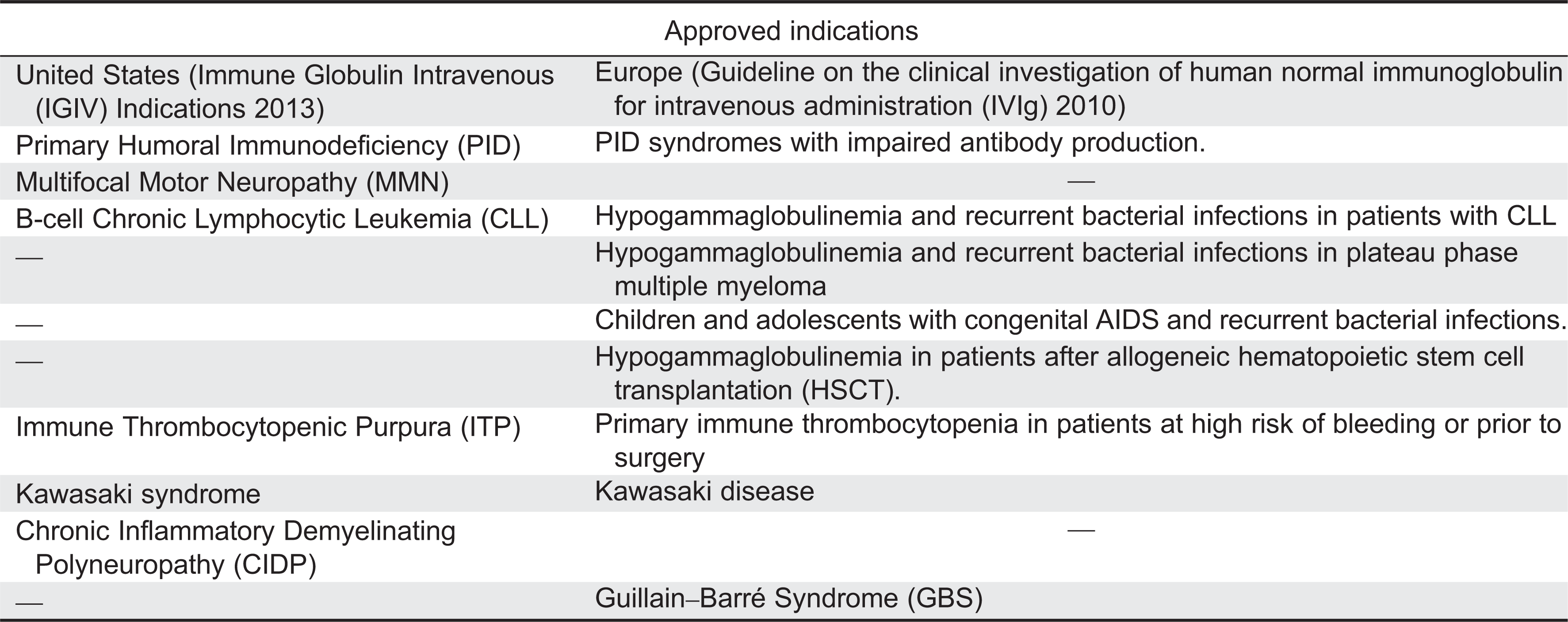
IVIG for antibody deficiencies
A principal use of immunoglobulin is to provide antibodies against infectious organisms to patients who are unable to make antibodies (Schiff 1994). Such patients include those with primary antibody deficiencies as well as patients with age-related antibody deficiencies.
Neonates
Because premature and low birth weight (LBW) infants are deficient in IgG, intervention with IVIG has been studied extensively. Nosocomial infections are a significant cause of morbidity and mortality in premature and LBW infants. A 25% incidence of late-onset infection was reported in a cohort of 6911 very LBW infants who were admitted to 12 US centres and who survived beyond 3 days (Stoll et al. 1996). The neonates who developed late-onset sepsis were significantly more likely to die than those who were not infected (17% vs 7%; p < 0.0001) (Stoll et al. 1996).
Baker et al. (1992) conducted a multicentre, double-blind study of 588 neonates weighing 500–1750 g at birth. The neonates were randomized to receive infusions of 500 mg/kg IVIG or placebo. IVIG was administered at enrollment (age 3–7 days), 1 week later, and then every 14 days thereafter until they had been given 5 IVIG infusions or had been discharged. There was a significant reduction in the risk of first nosocomial infection in IVIG recipients compared with placebo recipients. IVIG recipients with infections had fewer hospitalization days than control patients (80 days vs 101 days, p = 0.02) (Baker et al. 1992).
Fanaroff et al. (1994) studied 2416 very LBW infants weighing 501–1500 g in a multicentre controlled clinical trial of IVIG. Patients received their first dose of IVIG or placebo within 24 h of randomization. Patients who weighed between 501 and 1000 g were given 900 mg/kg IVIG and those who weighed between 1001 and 1500 g received 700 mg/kg IVIG. The mean number of infusions was 3. In this study, there was no significant difference in the incidence of infections between patients treated with IVIG compared with controls (Fanaroff et al. 1994).
In a review of 19 IVIG studies of 5000 preterm or LBW neonates treated prophylactically with IVIG, there was a statistically significant reduction in 1 or more episodes of sepsis and of any serious infection. There were no statistically significant differences in mortality or length of hospitalization (Ohlsson and Lacy 2013).
Chronic lymphocytic leukemia (CLL) and multiple myeloma (MM)
Bacterial infections are a major cause of morbidity and mortality in patients with CLL. The pathogenesis of infections is related to impaired humoral immunity caused by the disease and by therapy-related immunosuppression. Hypogammaglobulinemia is an important predisposing factor for infection. Suppression of cellular immunity with purine analogues such as fludarabine and monoclonal antibodies such as rituximab and alemtuzumab has introduced a new spectrum of infections by pathogens such as Pneumocystis, Listeria, mycobacteria, herpes viruses, Candida, and Aspergillus (Morrison 2010).
A randomized, double-blind, placebo-controlled, multicentre study of IVIG was conducted in CLL patients with either an IgG level ≤50% of the lower limit of normal or a history of 1 or more serious bacterial infections. Eighty-four patients were randomized to receive 400 mg/kg of IVIG or an equivalent volume of 0.9% sodium chloride every 3 weeks for 12 months. IVIG recipients had significantly fewer bacterial infections during the study than placebo recipients (23 vs 42, p = 0.01). IVIG recipients who were treated for a full year demonstrated a more striking reduction of bacterial infections (14 vs 36, p = 0.001) (Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia 1988).
Raanani et al. (2009) performed a systematic review and meta-analysis of 9 randomized, controlled trials that compared prophylaxis with IVIG versus a control in patients with CLL and MM. The primary outcomes were all-cause mortality and major infections. Three trials in CLL patients had evaluable outcome data on infections and mortality. The rate of “major” infections that required in-patient treatment and intravenous antibiotics was significantly lowered by IVIG prophylaxis but there was no significant effect on overall mortality. In the trial of multiple myeloma patients with outcomes included in the meta-analysis, IVIG had no effect on survival, but significantly reduced the incidence of serious bacterial infections (p = 0.002) and reduced the risk of recurrent infections. Interpretation of the data is complicated by the fact that the reviewed studies were relatively old (1988–1996). Since then, therapeutic protocols have become more immunosuppressive and management and outcomes have changed (Raanani et al. 2009).
HIV Infection
Treatment of children infected with HIV-1 began in 1979, before the virus had been identified. The children presented symptoms that were typical of a B-cell deficiency: recurrent bacterial infections, multiple septic episodes with the same organism, and failure to mount an antibody response to polysaccharide antigens (Deener et al. 2008). Treatment of the children with 300 mg/kg IVIG every 2 weeks abolished the recurrent bacterial infections and decreased the level of markers of immune activation such as neopterin and tumor necrosis factor (Deener et al. 2008). In a 10-year follow-up of 112 children, ages 9 months to 6 years treated with this regimen, there were no episodes of sepsis and only mild upper respiratory infections. The introduction of highly active antiretroviral therapies (HAART) has produced significant immune restoration of HIV-1 infected patients and has dramatically reduced the mortality and morbidity of these patients. No controlled studies have documented the effect of IVIG on patients who are infected with HIV-1 and are on HAART. Thus the role of IVIG in the post-HAART era is controversial (Deener et al. 2008).
Transplantation
Patients who undergo marrow or peripheral stem-cell transplantation are severely immune suppressed and are susceptible to infection. The use of IVIG in marrow transplant recipients was studied extensively in the 1980s and 1990s. In 1990, the National Institutes of Health consensus panel reviewed the clinical data of IVIG treatment of marrow transplant patients from controlled studies and noted that patients treated with 500 mg to 1 g IVIG weekly showed a decreased rate of acute graft versus host disease (GVHD) and a reduced rate of septicemia and local infection (NIH Consensus Dev Conf Statement 1990). On the basis of these studies, the use of IVIG for allogeneic marrow transplantation became a licensed indication in the United States.
A recent meta-analysis suggested that IVIG does not increase survival, prevent clinically documented infections, or decrease the risk of acute GVHD, but it does reduce the incidence of interstitial pneumonitis (Raanani et al. 2009). An increased risk of developing hepatic veno-occlusive disease was reported (Raanani et al. 2009).
IVIG as an immunomodulating agent
Kawsaki disease (KD)
Following the discovery that IVIG caused a rise in platelet counts in ITP patients, IVIG has been shown to be effective in suppressing inflammatory and autoimmune disorders. A successful application has been in the treatment of KD, which is an acute systemic vasculitis that progresses to cause coronary artery abnormalities in 25% of untreated patients (Jamieson and Singh-Grewal 2013). It is the leading cause of acquired cardiovascular disease in children in the developed world. Patients are predominantly male with approximately 75% of cases occurring in children between the ages of 3 months and 5 years. There is an increased incidence in people of Asian descent. The etiology is largely unknown but exposure to 1 or more environmental triggers is suspected (Jamieson and Singh-Grewal 2013).
The acute phase of KD lasts 1–2 weeks if untreated and is characterized by spiking a 40 °C fever and may present with cardiac manifestations including valvitis, pericarditis, and myocarditis (Jamieson and Singh-Grewal 2013). The efficacy of IVIG as the first-line treatment for acute phase KD has been proven in several clinical trials (Jamieson and Singh-Grewal 2013). The American Heart Association recommends that KD patients be treated with a single infusion of IVIG over 12 h at a dose of 2 g/kg within 10 days of fever onset along with 100 mg/kg per day of aspirin spread out over 4 doses until the child is afebrile (Jamieson and Singh-Grewal 2013). A recent Japanese trial found that the addition of 2 mg/kg prednisolone to the standard IVIG regiment significantly reduced adverse coronary outcomes (Jamieson and Singh-Grewal 2013)
Neurological diseases
The most recent clinical indications approved by regulatory agencies for IVIG are the treatment of patients with neurological conditions. Table 3 lists 3 neurological disorders approved in the United States or Europe for treatment with IVIG: multifocal motor neuropathy (MMN), Chronic inflammatory demyelinating polyneuropathy (CIDP), and Guillain–Barré Syndrome (GBS).
GBS is a subacute inflammatory demyelinating polyneuropathy that leads to severe quadriparesis (Schiff 1994). In the Netherlands, a large, multicentre trial was conducted in 147 patients who were randomly assigned to receive 5 sessions of plasma exchange or 400 mg/kg IVIG for 5 days (Van der Meché et al. 1992). Thirty-four percent of plasma exchange recipients improved 1 or more functional grades compared with 53% in the IVIG group. The median time to improvement was significantly shorter in the IVIG group (p = 0.05). Two smaller studies confirmed these results but 2 studies in the United States reported poor responses (Schiff 1994).
In 2008 Gamunex IVIG was approved for the treatment of CDIP. CIDP can be considered the chronic form of GBS (Schiff 1994). In a double-blind, placebo-controlled trial, 117 patients were randomized to receive 2 g/kg IVIG or an equal volume of placebo over 2–4 consecutive days. Maintenance doses of 1 g/kg or placebo were given every 3 weeks for up to 24 weeks (Hughes et al. 2008). Thirty-two of 59 subjects treated with IVIG (54%) responded to IVIG compared with 12 of 58 subjects (21%) administered placebo (p = 0.0002). During an extension phase, patients who continued to receive IVIG had a longer time to relapse than placebo recipients (p = 0.11) (Hughes et al. 2008).
In 2012, Gammagard Liquid IVIG was approved to treat patients with MMN. MMN is a rare immune-mediated disease that presents with distal motor weakness in one or more limbs and may produce functional impairment (Gammagard Liquid Prescribing information, 2012). Forty-four adult subjects were enrolled in a double-blind, placebo-controlled, cross-over study that examined the grip strength in the more affected hand and the disability of the upper limb. IVIG produced a significantly reduced decline in mean grip strength than placebo. Progression of upper limb clinical disability was also significantly reduced by IVIG compared with placebo (Gammagard Liquid Prescribing information 2012).
Subcutaneous immunoglobulin therapy
Berger et al. (1980) published a paper on the use of small pumps to infuse patients with ISG subcutaneously at rates of 1–2 mL/h instead of by intramuscular injection. Termed “slow subcutaneous infusions,” the infusions were accompanied with little swelling or pain at the infusion site and no systemic reactions (Berger et al. 1980).
Subcutaneous infusions of immunoglobulin have become a popular mode of delivery in patients with primary immunodeficiency. Gardulf et al. (1991) reported on a clinical trial of rapid subcutaneous immunoglobulin infusions in 25 consecutive patients with hypogammaglobulinemia who had been previously treated with intramuscular ISG injections. Patients were infused at 34–40 mL/h with 100 mg/kg per week with a mercury-free ISG (produced by Kabi) delivered in divided doses at 2–6 infusion sites on the abdominal wall and (or) the thigh. Patients were trained at the study site to infuse themselves and after 6 months of treatment received their remaining infusions at home. The authors reported that the subjects experienced fewer adverse reactions compared with ISG treatment (Gardulf et al. 1991). Since this article was published, there have been many follow-up publications by the same authors and subcutaneous immunoglobulin therapy has become a standard treatment for patients with immunodeficiency in Scandinavian countries (Lingman-Framme and Fasth 2013).
In 2006, the safety and efficacy of a mercury-free ISG preparation (Vivaglobin, CSL Behring) was studied in an open-label multicentre study in PID patients in the United States (Ochs et al. 2006). Sixty-five patients previously treated with IVIG were enrolled in a 3-month SCIG treatment period. Fifty-one of these patients were then treated for 12 months with weekly SCIG infusions at a dose calculated to be equal to 137% of their previous IVIG dose. This resulted in a mean weekly dose of 158 mg/kg per week with a range of 155–165 mg/kg per week. Trough IgG levels increased from 786 mg/dL at baseline to 1040 mg/dL during SCIG treatment. The first 2 subcutaneous infusions were administered under the supervision of trained nurses. SCIG was administered at 1 or more injection sites using an infusion pump. Infusion rates were ≤ 20 mL/h at any single injection site and the volume per site was ≤15 mL (Ochs et al. 2006).
During the 12-month efficacy phase, 2 serious bacterial infections (SBI) were reported, both pneumonias, for an incidence of 0.04 SBI per patient per year. The annual rate of all infections was 4.43 per patient per year and the number of days with infection was 118 per patient-year. The most frequent AE was infusion site reaction, which occurred at least once in 60 patients (91%). The next most frequent AE was headache. Thirty-one subjects (48%) reported a total of 159 episodes of headache during the 12-month efficacy period (Ochs et al. 2006).
The same manufacturer recently licensed another SCIG known by the trade name Hizentra®. Hizentra is a 20% (w/v) IgG solution that was studied in an open-label, multicentre, single-arm study of safety and efficacy (Hagan et al. 2010). Initially, 38 patients were treated for 12 weeks with a weekly dose that was calculated from the last 3 IVIG infusions received prior to the study and multiplied by 1.30 as reported previously (Ochs et al. 2006). The mean dose during this phase ranged from 176.8 to 182.9 mg/kg per week. During the efficacy period that followed, 23 subjects received 54 SCIG infusions, and 15 subjects received from 11 to 53 SCIG infusions at mean doses of 179.6–224.3 mg/kg per week (Hagan et al. 2010). The median IgG trough level during this period was 12.5 ± 3.2 mg/mL.
There were no serious bacterial infections during the study. There were 96 nonserious bacterial infections in 31 patients. Sinusitis was the most common infection followed by nasopharyngitis. Twelve patients missed 71 days of work or school (Hagan et al. 2010). All 49 patients experienced local AEs. Excluding local reactions, 45 patients experienced at least 1 AE and 25 patients had an AE that was considered to be related to the study product (Hagan et al. 2010). Results of these studies are listed in Table 4.
Table 4:
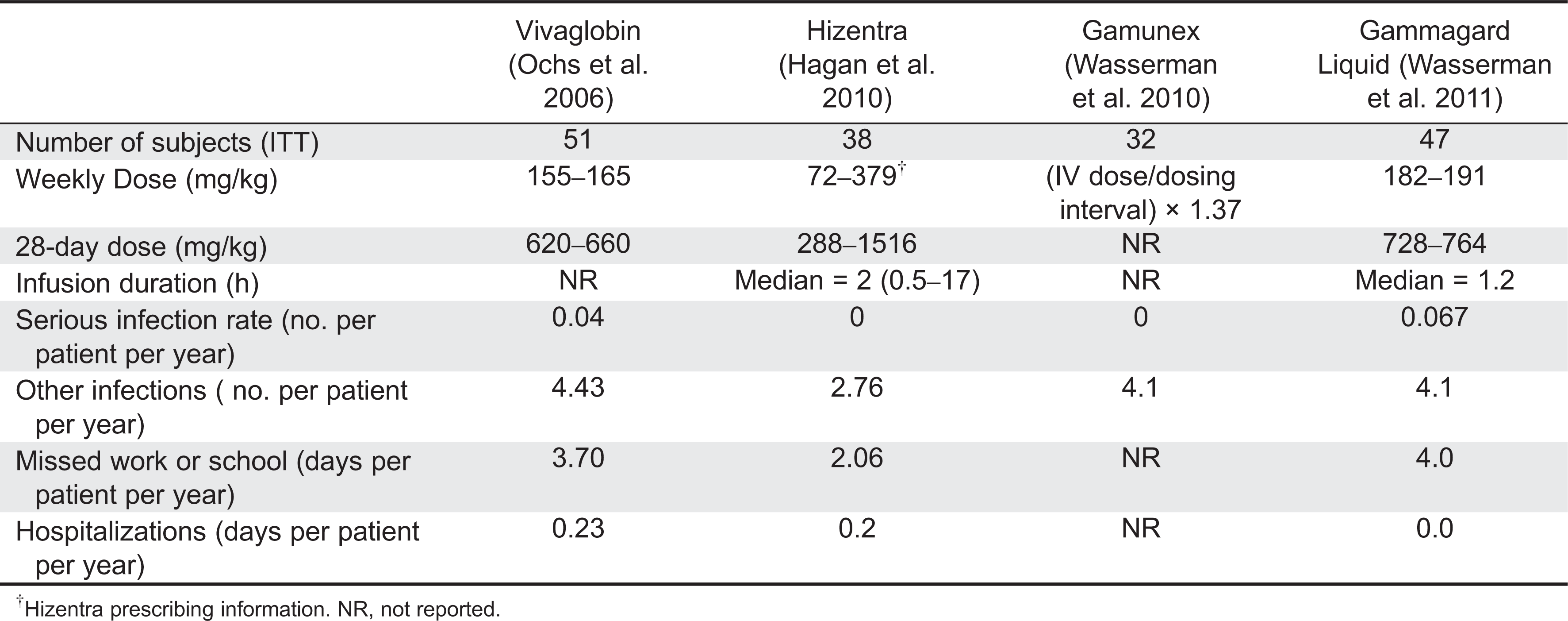
Two IVIG preparations have been approved for subcutaneous (SC) administration: Gamunex and Gammagard Liquid. Gamunex was studied in an open-label pharmacokinetic trial to compare the steady state area under the concentration–time curve (AUC) of subcutaneous infusions compared with the AUC obtained from intravenous infusions (Wasserman et al. 2010). All patients received IV infusions of Gamunex for 3–4 months prior to being switched to weekly SC Gamunex infusions. The SC dose was determined by dividing the IV dose by the dosing interval (3 or 4 weeks) and multiplying by 1.37. The mean adjusted AUC0-t values for SC and IV administration were 6858 mg·h/mL and 7640 mg·h/mL, respectively. Statistical analysis of these values showed that SC Gamunex was not inferior to IV Gamunex. The mean overall steady-state trough concentration of total plasma IgG was 11.4 mg/mL during SC administration and 9.6 mg/mL following IV administration (Wasserman et al. 2010). Other results are shown in Table 4.
Subcutaneous administration of Gammagard Liquid was also studied in an open-label trial of 49 patients with PID (Wasserman et al. 2011). All subjects received IV infusions of Gammagard Liquid for 13 weeks. One week after the final IV dose, the subjects began to receive weekly subcutaneous doses at 137% of the IV dose. The plasma IgG trough level was 12.0 ± 2.8 mg/mL during SC administration and 10.5 ± 2.60 during IV treatment. AUC values were 9176 ± 1928 days × mg/dL for SC treatment and 9958 ± 2274 days × mg/dL for IV treatment, and were determined to be equivalent. Additional safety and efficacy results are shown in Table 4.
In summary, SCIG clinical trials have shown relatively high IgG trough levels when compared with IVIG in the same study (Wasserman 2010, 2011).
SCIG clinical applications
In the United States, primary humoral immunodeficiency is the only approved indication for SCIG and subcutaneously administered IVIG. Other clinical applications have been reported in the literature. In the United Kingdom, SCIG has been used to treat 17 patients with autoimmune neuropathies and 1 patient with Epidermolysis bullosa acquista (Misbah et al. 2011). A retrospective study of 12 children with prolonged hypogammaglobulinemia after hematopoietic stem cell transplantation found that they experienced fewer side-effects and no more infections that children who received IVIG (Lingman-Framme and Fasth 2013). SCIG has also been used instead of IVIG in maintenance therapy of patients with MMN and CDIP (Jolles et al. 2011). SCIG therapy has also been studied as the first immunoglobulin treatment of patients with polymyositis or dermatomyositis (Jolles et al. 2011).
It is noteworthy that the first patient with primary immunodeficiency treated by Bruton (1952) was administered immunoglobulin subcutaneously. SCIG has become increasingly popular in the treatment of patients with PID and, as new clinical trials are conducted, may become an alternative to IVIG for other clinical indications.
AEs of immunoglobulins
Clinical trials data for both IVIG and SCIG are relatively standardized with respect to the reporting of AEs since the FDA guidance was issued (FDA/CBER Guidance for Industry 2008). A review of FDA approved labeling of licensed immunoglobulins indicates that the most common temporally associated (within 72 h) AE associated with immunoglobulins is headache, which occurs at a frequency of 8%–15% of IVIG infusions. Headache is also observed in SCIG recipients at rates of 1.4%–4%. Other adverse events experienced by IVIG recipients are fatigue, fever, and nausea. SCIG recipients experience the same adverse events and also local adverse events such as discomfort and pain, erythema, swelling/edema, and pruritis (Biotest 2012; Baxter 2013, 2014; CSL Behring 2015).
REFERENCES
Amman A.J., Ashman R.F., Buckley R.H., Hardie W.R., Krantmann H.J., Nelson J., Ochs H., Stiehm E.R., Tiller T., Wara D.W., and Wedgwood R. Use of intravenous gamma-globulin in antibody immunodeficiency: Results of a multicenter controlled trial Clin. Immunol. Immunopathol 1982 22 1 60 -67
Baker C.J., Melish M.M., Hall R.T., Casto D.T., Vasan U., and Givner L.B. Intravenous immune globulin for the prevention of nosocomial infection in low-birth-weight neonates N. Engl. J. Med 1992 327 4 213 -219
Barandun S., Kistler P., Jeunet F., and Isliker H. Intravenous administration of human gamma globulin Vox Sang 1962 7 157 -174
Baxter Healthcare Corporation, Westlake Village, CA. Gammagard Liquid [Immune globulin infusion (Human) 10%], Prescribing information, 2013.
Baxter Healthcare Corporation. HYQVIA [Immune Globulin Infusion (Human) 10% with Recombinant Human Hyaluronidase] Prescribing information, 2014.
Bender S. and Hetherington S. Haemophilus influenzae type b opsonins of intravenous immunoglobulins J. Clin. Immunol 1987 7 6 475 -480
Berger M., Cupps T.R., and Fauci A.S. Immunoglobulin replacement therapy by slow subcutaneous infusion Ann Int Med 1980 93 55 -56
Berger M., Pinciaro P.J., Althaus A., Ballow M., Chouksey A., Moy J., Ochs H., and Stein M. Efficacy, pharmacokinetics, safety, and tolerability of Flebogamma® 10% DIF, a high-purity human intravenous immunoglobulin, in primary immunodeficiency J. Clin. Immunol 2010 30 2 321 -329
Bernatowska E., Madalinski K., Janowicz W., Weremowicz R., Gutkowski P., Wolf H.M., and Eibl M.M. Results of a prospective controlled two-dose crossover study with intravenous immunoglobulin and comparison (retrospective) with plasma treatment Clin. Immunol. Immunopathol 1987 43 2 153 -162
Biotest Pharmaceuticals Corporation. BIVIGAM [Immune Globulin Intravenous (Human), 10% Liquid, Prescribing Information, 2012. Biotest, Boca Raton, FL.
Bruton O.C. Agammaglobulinemia Pediatrics 1952 9 722 -727
Bussel JB, Kimberly RP, Inman R.D., Schulman I., Cunningham-Rundles C., Cheung N., Smithwick E.M., O’Malley J., Barandun S., and Hilgartner M.W. Intravenous gammaglobulin treatment of chronic idiopathic thrombocytopenic purpura Blood 1983 62 480 -486
Cohn E.J., Oncley J.L., Strong L.E., Hughes W.L. Jr., and Armstrong S.H. Jr Chemical, clinical, and immunological studies on the products of human plasma fractionation. I. The characterization of the protein fractions of human plasma J. Clin. Invest 1944 23 4 417 -432
Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia N. Engl. J. Med 1988 319 14 902 -907
CSL Behring AG, HIZENTRA [Immune Globulin Subcutaneous (Human) 20% Liquid, Prescribing Information, 2015.
Deener A., Mehra A., Bernstein L., Shliozberg J., and Rubinstein A. Intravenous gammaglobulin treatment in HIV-1 infection Immunol. Allergy. Clin. N. Am 2008 28 4 851 -859
Eijkhout H.W., van Der Meer J.W., Kallenberg C.G., Weening R.S., van Dissel J.T., Sanders L.A., Strengers P.F., Nienhuis H., and Schellekens P.T. Inter-University Working Party for the Study of Immune Deficiencies. The effect of two different dosages of intravenous immunoglobulin on the incidence of recurrent infections in patients with primary hypogammaglobulinemia. A randomized, double-blind, multicenter crossover trial Ann Intern Med 2001 135 3 165 -74
Encyclopædia Britannica. 2014. Robert Koch. Encyclopædia Britannica Online. Encyclopædia Britannica Inc. <http://www.britannica.com/EBchecked/topic/320834/Robert-Koch>.
Enders J.F. Chemical, clinical, and immunological studies on the products of human plasma fractionation. X. The concentrations of certain antibodies in globulin fractions derived from human blood plasma J. Clin. Invest 1944 23 4 510 -530
Fanaroff A.A., Korones S.B., Wright L.L., Wright E.C., Poland R.L., Bauer C.B., Tyson J.E., Philips J.B., Edwards W., Lucey J.F., Catz C.S., Shankaran S., and Oh W. A controlled trial of intravenous immune globulin to reduce nosocomial infections in very-low-birth-weight infants N. Engl. J. Med 1994 330 16 1107 -1113
FDA Blood Products Advisory Committee Meeting. March 1999. Transcript. http://www.fda.gov/ohrms/dockets/ac/99/transcpt/3504t2.pdf.
FDA Blood Products Advisory Committee Meeting. March 2000. Transcript. http://www.fda.gov/ohrms/dockets/ac/00/transcripts/3603t2a.pdf.
FDA/CBER Guidance for Industry. 2005. Safety, efficacy, and pharmacokinetic studies to support marketing of immune globulin intravenous (human) as replacement therapy for primary humoral immunodeficiency. http://www.fda.gov/cber/guidelines.htm.
FDA/CBER Guidance for Industry. 2008. Safety, efficacy, and pharmacokinetic studies to support marketing of immune globulin intravenous (human) as replacement therapy for primary humoral immunodeficiency. http://www.fda.gov.cber/gdlns/igivimmuno.htm.
Gardulf A., Hammarström L., and Smith C.I.E. Home treatment of hypogammaglobulinaemia with subcutaneous gammaglobulin by rapid infusion Lancet 1991 338 162 -166
Guideline on the clinical investigation of human normal immunoglobulin for intravenous administration (IVIg). 2010. EMA/CHMP/BPWP/94033/2007 rev. 2.
Hagan J.B., Fasano M.B., Spector S., Wasserman R.L., Melamed I., Rojavin M.A., Zenker O., and Orange J.S. Efficacy and safety of a new 20% immunoglobulin preparation for subcutaneous administration, IgPro20, in patients with primary immunodeficiency J. Clin. Immunol 2010 30 5 734 -745
Hagenbeek A., Brummelhuis G.J., Donkers A., Dumas A.M., Haaft A.T., Schaap B.J.P., Sizoo W., and Lowenberg B. Rapid clearance of cytomegalovirus-specific IgG after repeated intravenous infusions of human immunoglobulin into allogeneic bone marrow transplant recipients J. Infect. Dis 1987 155 5 897 -902
Heidelberger M. and Pedersen K.O. The molecular weight of antibodies J. Exp. Med 1937 65 3 393 -414
Hughes R.A., Donofrio P., Bril V., Dalakas M.C., Deng C., Hanna K., Hartung H.-P., Latov N., Merkies I.S., and Van Doorn P.A. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomised placebo-controlled trial Lancet. Neurol 2008 7 2 136 -144
Imbach P., Barandun S., Baumgartner C., Hirt A., Hofer F., and Wagner H.P. High-dose intravenous gammaglobulin therapy of refractory, in particular idiopathic, thrombocytopenia in childhood Helv. Paediat. Acta 1981 46 81 -86
Immune Globulin Intravenous (IGIV) Indications. 2013. www.fda.gov/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/LicensedProductsBLAs/FractionatedPlasmaProducts/ucm133691.htm.
Jamieson N. and Singh-Grewal D. Kawasaki disease: A clinician’s update Int. J. Pediatrics 2013 2013 1 -7
Janeway C.A., Apt L., and Gitlin D. Agammaglobulinemia Trans. Assoc. Am. Phy 1953 66 200 -202
Janeway C.A., Merler E., Rosen FS, Salmon S., and Crainet J.D. Intravenous gamma globulin. Metabolism of gamma globulin fragments in normal and agammaglobulinemic persons N. Engl. J. Med 1968 278 919 -923
Jolles S., Sein M., Longhurst H.J., Borte M., Ritchie B., Sturzenegger M. H., and Berger M. New frontiers in subcutaneous immunoglobulin treatment Biol. Ther 2011 1 1 1 -15
Kim K.S., Wass C.A., Kang J.H., and Anthony B. Functional activities of various preparations of human intravenous immunoglobulin against type III group B Streptococcus J. Infect. Dis 1986 153 6 1092 -1097
Kitasato S. Über dem Tetanusbacillus Zeitschrift fur Hygience und Infektionskrankheiten 1889 7 225 -234
Lingman-Framme J. and Fasth A. Subcutaneous immunoglobulin for primary and secondary immunodeficiencies: An evidence-based review Drugs 2013 73 12 1307 -1319
McKhann C.F. and Chu F.T. Antibodies in placental extracts J. Infect. Dis 1933 52 2 268 -277
Medical Research Council Hypogammaglobulinemia in the United Kingdom Lancet 1969 1 163 -168
Misbah S., Kuijpers T., van der Heijden Grimbacher B., Guzman D., and Orange J.J. Bringing immunoglobulin knowledge up to date: how should we treat today? Current knowledge in immunoglobulin therapy Clin. Exp. Immunol 2011 166 1 16 -25
Morrison V.A. Infectious complications of chronic lymphocytic leukemia: pathogenesis, spectrum of infection, preventive approaches Best. Pract. Res. Clin. Haematol 2010 23 1 145 -153
Moy J.N., Scharenberg A.M., Stein M.R., Suez D., Roberts R.L., Levy R.J., Ballow M., Fasano M.B., Dash C.H., and Leach S.J. Efficacy and safety of a new immunoglobulin G product, Gammaplex®, in primary immunodeficiency diseases Gammaplex® in PID Clin. Exp. Immunol 2010 162 3 510 -515
NIH Consensus Dev Conf Statement 1990 May 21–23; 8(5). Intravenous immunoglobulin: Prevention and treatment of Disease.
Nolte M.T., Pirofsky B., Gerritz G.A., and Golding B. Intravenous immunoglobulin therapy for antibody deficiency Clin. Exp. Immunol 1979 36 237 -243
Ochs H.D., Gupta S., Kiessling P., Nicolay U., and Berger M. Subcutaneous IgG study group. Safety and efficacy of self-administered subcutaneous immunoglobulin in patients with primary immunodeficiency diseases J. Clin. Immunol 2006 26 3 265 -273
Ohlsson A. and Lacy J.B. Intravenous immunoglobulin for preventing infection in preterm and/or low birth weight infants Cochrane Database Systemat. Rev 2013 7 Art. No.: CD000361
Pollack M. Antibody activity against Pseudomonas aeruginosa in immune globulins prepared for intravenous use in humans J. Infect. Dis 1983 147 6 1090 -1098
Raanani P., Gafter-Gvili A., Paul M.M., Ben-Bassat I., Leibovici L., and Shpilberg O. Immunoglobulin prophylaxis in chronic lymphocytic leukemia and multiple myeloma: systematic review and meta-analysis Leukemia Lymphoma 2009 50 5 764 -772
Raanani P., Gafter-Gvili A., Paul M., Ben-Bassat I., Leibovici L., and Shpilberg O. Immunoglobulin prophylaxis in Hematopoietic stem cell transplantation: Systematic review and meta-analysis J. Clin. Oncol 2009 27 5 770 -781
Roifman C.M., Lederman H.M., Lavi S., Stein L.D., Levison H., and Gelfand E.W. Benefit of intravenous IgG replacement in hypogammaglobulinemic patients with chronic sinopulmonary disease Am. J. Med 1985 79 2 171 -174
Roifman C.M., Levison H., and Gelfand E.W. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinemia and chronic lung disease Lancet 1987 1 8541 1075 -1077
Schiff R.I. Intravenous gammaglobulin: pharmacology, clinical uses and mechanisms of action Pediatr. Allergy. Immunol 1994 5 2 63 -87
Schroeder D.D., Tankersley D.L., and Lundblad J.L. A new preparation of modified immune serum globulin (human) suitable for intravenous administration I: Standardization of the reduction and alkylation reaction Vox. Sang 1981 40 6 373 -382
Schultze H.E. and Schwick G. Über neue Möglichkeiten intravenöser Gammaglobulin-Applikation Deuthsche. Med. Wchnschr 1962 87 34 1643 -1650
Sgouris J.T. The preparation of plasmin-treated immune serum globulin for intravenous application Vox. Sang 1967 13 71 -84
Sri Kantha S.A. Centennial review; the 1890 tetanus antitoxin paper of von Behring and Kitasato and the related developments Keio. J. Med 1991 40 1 35 -39
Steele R.W. and Steele R.W. Functional capacity of immunoglobulin G preparations and the F(ab′)2 split product J. Clin. Microbiol 1989 27 640 -664
Stein M.R., Nelson R.P., Church J.A., Wasserman R.L., Borte M., Vermylen C., and Bichler J. Safety and efficacy of Privigen®, a Novel 10% liquid immunoglobulin preparation for intravenous use, in patients with primary immunodeficiencies J. Clin. Immunol 2009 29 1 137 -144
Stokes J., Maris E.P., and Gellis S.S. Chemical, clinical, and immunological studies on the products of human plasma fractionation. XI. The use of concentrated normal human serum gamma globulin (human immune serum globulin) in the prophylaxis and treatment of measles J. Clin. Invest 1944 23 4 531 -540
Stoll B.J., Gordon T., Korones S.B., Shankaran S., Tyson J.E., Bauer C.R., Fanaroff A.A., Lemons J.A., Donovan E.F., Oh W., Stevenson D.K., Ehrenkranz R.A., Papile L.-A., Verter J., and Wright L.L. Late onset sepsis in very low birth weight neonates: A report from the national institute of child health and human development neonatal research network J. Pediatrics 1996 129 1 63 -71
Tiselius A. Electrophoresis of purified antibody preparations J. Exp. Med 1937 65 5 641 -646
Tiselius A. and Kabat E.A An electrophoretic study of immune sera and purified antibody preparations J. Exp Med 1939 69 1 119 -131
Turner, M. 1989. Molecules which recognize antigen. In Immunology. Edited by IM Roitt, J Brostoff, DK Male. CV Mosby, St. Louis, MO. p 5.8.
Ullmann A. Pasteur-Koch: Distinctive ways of thinking about infectious diseases Microbe 2007 2 8 383 -387
Van der Meché F.G.A., Schmitz P.I.M., and The Dutch Guillain—Barré Study Group A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome New. Eng. J. Med 1992 326 17 1123 -1129
von Behring E. Untersuchungen über das Zustandekommen der Diphtherie-Immunität bei Thieren Deutscbe. Med. Wochenschr 1890 16 50 1145 -1148
von Behring E. and Kitasato S. Über das zustandekommen der Diphtherie-Immunität und der Tetanus-Immunität bei Thieren Dtsch. med Wochenschr 1890 49 1113 -1114
Wasserman R.L., Church J.A., Stein M.R., Moy J., White M., Strausbaugh S., Schroeder H., Ballow M., Harris J., Melamed I., Elkayam D., Lumry W., Suez D., and Rehman S.M. Safety, efficacy, and pharmacokinetics of a new 10% liquid intravenous immunoglobulin (IVIG) in patients with primary immunodeficiency J. Clin. Immunol 2012 32 4 663 -669
Wasserman R.L., Irani A.M., Tracy J., Tsoukas C., Stark D., Levy R., Chen J., Sorrells S., Roberts R., and Gupta S. Pharmacokinetics and safety of subcutaneous immune globulin (human), 10% caprylate/chromatography purified in patients with primary immunodeficiency disease Clin. Exp. Immunol 2010 161 3 518 -526
Wasserman R.L., Melamed I., Kobrynski L., Strausbaugh S.D., Stein M.R., Sharkhawy M., Engl W., Leibl H., Sobolevsky L., Gelmont D., Schiff R.I., and Grossman W.J. Efficacy, safety, and pharmacokinetics of a 10% liquid immune globulin preparation (GAMMAGARD LIQUID, 10%) administered subcutaneously in subjects with primary immunodeficiency disease J. Clin. Immunol 2011 31 3 323 -331
Winston D.J., Ho W.G., Rasmussen L.E., Lin C.-H., Chu C.L., Merigan T.C., and Gale R.P. Use of intravenous immune globulin in patients receiving bone marrow transplants J. Clin. Immunol. 1982 2 April Supplement S2 42S -47S
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 4 • December 2015
Pages: 181 - 194
History
Received: 29 December 2014
Accepted: 26 February 2015
Accepted manuscript online: 7 April 2015
Version of record online: 7 April 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
John A. Hooper. 2015. The history and evolution of immunoglobulin products and their clinical indications. LymphoSign Journal.
2(4): 181-194. https://doi.org/10.14785/lpsn-2014-0025
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
