Immunodeficiencies with hypergammaglobulinemia: a review
Abstract
Primary immunodeficiencies (PID) can present with recurrent infections, autoimmunity, inflammation, or malignancy and each of these conditions can be associated with elevated immunoglobulin. A high level of immunoglobulin G (IgG) is an uncommon finding, especially in pediatrics, and does not rule out primary immunodeficiency. Deficiencies in varied aspects of immune response have been described with high IgG. Reported PID conditions with elevated IgG include defects in humoral, cellular, and innate immunity. Some of these immunodeficiencies can have fatal outcomes, some require hematopoetic stem cell transplantation, and some require systemic medications. The mechanisms driving elevated IgG are not well understood, but in some cases abnormal cytokine production has been proposed. The evaluation of a patient with high IgG is guided by the patient's history and a physical examination, with special attention to autoimmunity in pediatrics and malignancy and liver disease in adults. In the setting of autoimmunity, chronic gastrointestinal disease, or chronic infections, the measurement of specific antibodies to evaluate the function of the IgG should be considered. An increased appreciation of elevation in IgG reflecting immune dysregulation may lead to earlier PID diagnoses.
Introduction
In the course of a work-up for symptoms such as recurrent fevers and infections or concern about autoimmunity, the patient's level of IgG is often tested. Multiple papers and guidelines exist to assist the clinician with the immunodeficiency differential diagnosis for hypogammaglobulinemia, but high serum IgG has not been characterized in detail.
Differential diagnosis of high IgG
One of the largest retrospective series of mostly adult patients (aged 15–80 years) with elevated immunoglobulin (Dispenzieri et al. 2001) discovered that patients with high IgG fall into 1 of 5 major categories: liver disease, connective tissue diseases, chronic infections, hematologic disorders, and nonhematologic malignancies. No patients with immunodeficiency were described in this series or in the tables describing a wide variety of conditions with high IgG. Interestingly, 1 patient without a diagnosis did have poorly defined inflammatory lung disease with bronchiectasis and idiopathic pulmonary fibrosis, raising the possibility of immunodeficiency.
In the pediatric population, a recent retrospective study described the diagnoses of patients with IgG over 20 g/L (Lo et al. 2013). In this study, high IgG was associated with autoimmune disorders, especially lupus and arthritis, as well as infectious etiology, malignancy or stem cell transplantation, immunodeficiency, medications, and other conditions such as portal hypertension. The rarity of the finding of hypergammaglobulinemia in children was stressed by Lo et al. (2013) because during a decade at a tertiary children's hospital, only 442 patients were identified with high IgG. However, given their methodology, patients who were on intravenous immunoglobulin (IVIG) were understandably excluded. Therefore, the patients with diagnosed immunodeficiencies requiring immunoglobulin replacement were not included in this number. Only 15 of 442 patients had a recognized immunodeficiency. The captured immunodeficiencies with high IgG were varied and included hyper-IgE syndrome, ataxia–telangectasia, and autoimmune lymphoproliferative disease.
Primary immunodeficiencies with high immunoglobulins could be missed
Despite an effort to classify primary immunodeficiencies (PID) (Geha et al. 2007; Notarangelo et al. 2009), multiple recent publications have brought attention to the fact that PID with the same genetic mutation can have very variable presentations (Al-Herz and Notarangelo 2012; Maggina and Gennery 2013). It is already recognized that normal numbers of lymphocytes do not rule out immunodeficiency. For example, severe combined immunodeficiency (SCID) can present with a normal or elevated lymphocyte count due to either oligoclonal proliferation of T cells as seen in Omenn's syndrome (Villa et al. 2008) or expansion of the B-cell numbers in conditions such as JAK3 or IL-2 receptor gamma deficiency. Just as normal or elevated numbers of lymphocytes do not rule out immunodeficiency, the finding of elevated immunoglobulin may not be reassuring as to immune status. A long diagnostic delay for patients with common variable immunodeficiency has been recognized (Wood et al. 2007) and it is reasonable to assume that high immunoglobulins would delay diagnosis of an immunodeficiency even further.
Some immunodeficiencies are very well described as having high levels of immunoglobulins. For some conditions, the high immunoglobulin level is extremely informative as to the nature of the immunodeficiency. A collection of disorders have hyper-IgM as a result of the impaired ability of the B cell to switch immunoglobulin class (Ochs 2008). High IgE is a characteristic finding in hyper-IgE syndrome (Freeman and Holland 2009). Elevations in IgM and IgE are components of immunodeficiencies that are recognizable by that characteristic and will not be the focus of this review.
In contrast, hyper-IgG does not have a specific phenotype or allude to a specific diagnosis, but it includes very significant immunodeficiencies. This review aims to present the wide range of immunodeficiencies associated with hypergammaglobulinemia, including SCID, to illustrate that it is a described feature of many immunodeficiencies and to increase the recognition that the function of the IgG is more critical than the level.
An illustrative case
A 20-year-old male visited his primary care doctor because of recurrent sinus infections and 2 pneumonia diagnoses in the preceding year. His physical exam results were normal. Laboratory studies showed a normal blood count, and analysis of the immunoglobulins, performed to rule out hypogammaglobulinemia, showed elevated IgG. A serum protein electrophoresis that excluded monoclonality was performed. The evaluation was considered normal and immunodeficiency was considered to be ruled out. The elevated IgG was ascribed to the recurrent sinus infections.
Years later, a severe pneumonia prompted a referral to the Immunology Department. The Immunologist checked the function of the immunoglobulins by examining antibody titres. Specific antibody titres showed no response to protein and polysaccharide vaccinations despite an up-to-date schedule. By this point, a CT scan of the chest scan revealed bronchiectasis. A diagnosis of antibody deficiency with normal or elevated immunoglobulins (or dysgammglobulinemia) was made and the patient was started on intravenous gammaglobulin.
Immunglobulin G
Immunoglobulins are proteins composed of heavy chains and light chains. Their class is defined by the constant region of the heavy chain. IgG is the most abundant immunoglobulin and therefore constitutes most of the gamma region in serum protein electrophoresis. For the purposes of this review, hypergammaglobulinemia will focus on high IgG.
IgG is produced by B lymphoctytes. A naïve B cell can produce IgD and IgM, but help is required to produce IgG cognate T lymphocyte. The B cell must receive the appropriate signals from the T lymphocyte to make an irreversible change in its DNA to undergo class switching to produce IgG. Without these signals, or without T lymphocytes, IgG cannot be produced.
IgG has important roles in immunity including complement activation and opsonization. The function of IgG differs by subclass. IgG is classified into 4 subclasses. IgG1 is the most abundant subclass and makes up about two-thirds of the total IgG. IgG1 and IgG3 can recognize protein and viral antigens. IgG2 recognizes polysaccharide antigens. The function of IgG4 is less elucidated (Aalberse et al. 2009). IgG4 does not fix complement, and it increases in allergen immunotherapy and parasitic infections. The levels of IgG are age dependent (Schauer et al. 2003), so appropriate reference ranges should be used.
Immunodeficiencies with high IgG
A list of immunodeficiency conditions reported with hypergammaglobulinemia is presented in Table 1. This table highlights a wide range of immunodeficiencies, including fatal immunodeficiencies, that can have high IgG. They have been mostly organized according to the newest update on the classification of PIDs from the international union of immunological societies expert committee for PID (Al-Herz et al. 2014) in which the only PIDs specifically listed to be associated with high IgG are mammalian sterile20-like kinase 1 (MST1), also known as serine threonine kinase 4 (STK4) deficiency and autoimmune lymphoproliferative syndrome (ALPS), although many are described as having normal immunoglobulin levels. It is important to also recognize that many more of these PID conditions have been reported with elevated IgG to increase their recognition.
Table 1:
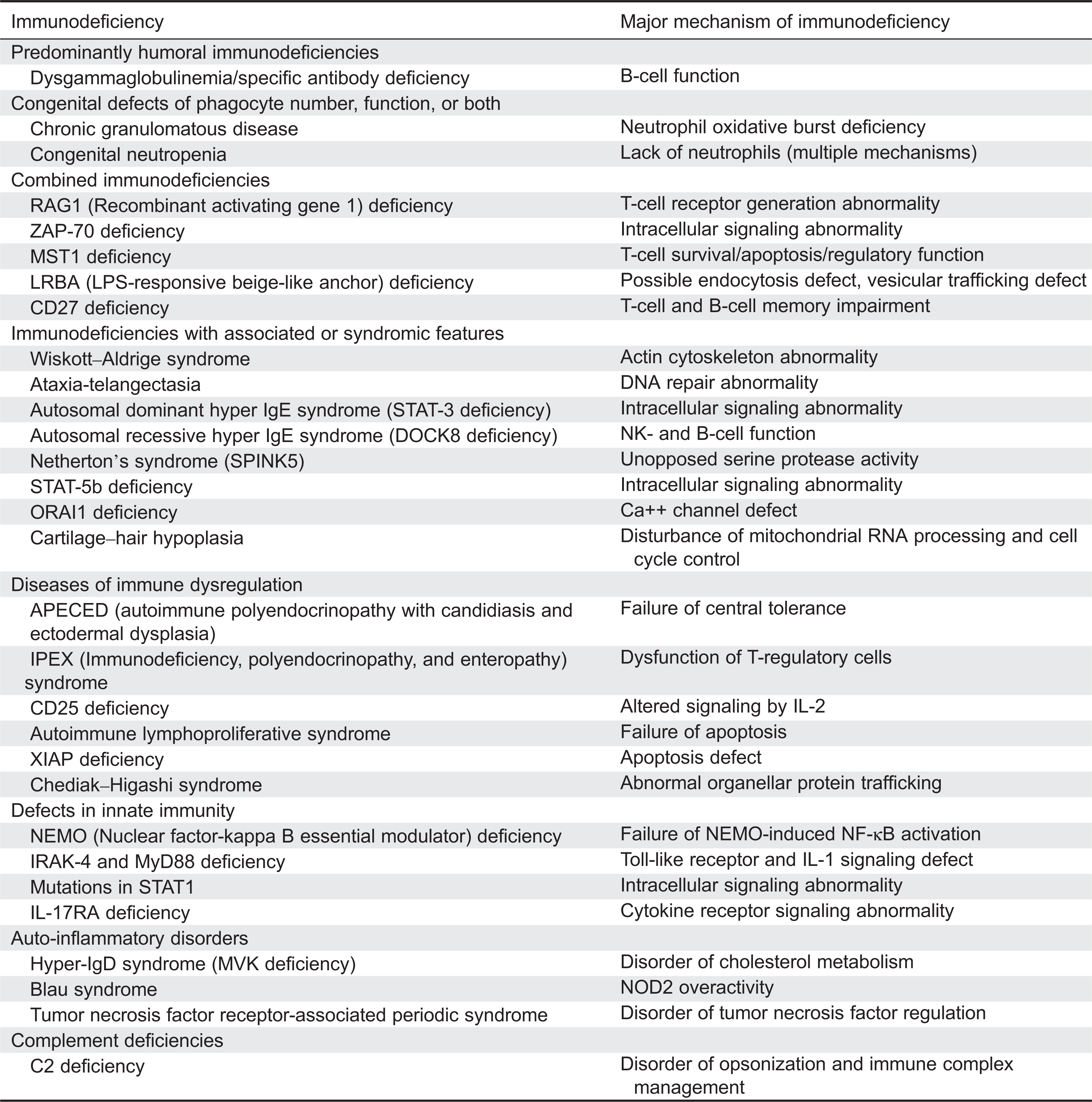
Hypergammaglobulinemia in congenital defects of phagocyte number or function or both
Since the 1950s, there has been recognition that significant immunodeficiencies can occur in the setting of high IgG. Chronic granulomatous disease (CGD) was first described as a disorder in which recurrent infections occur in the setting of hypergammaglobulinemia. This description predated the more advanced testing we have today and was intended to differentiate CGD from Bruton's agammaglobulinemia (Holland 2013). In CGD there is impairment in NADPH oxidase function. The neutrophils cannot generate superoxide anions and are therefore incapable of creating reactive oxygen intermediates and mounting an appropriate oxidative burst. This impairment leads to a susceptibility to infections with catalase-positive organisms. The NADPH oxidase also contributes to the formation of neutrophil extracellular traps (NETS), extracellular fibres released from the neutrophils that can bind pathogens. These patients can present with recurrent abscesses, Aspergillus infections, candidal infections, and Crohn's disease-like gastrointestinal disease. CGD can be inherited in an X-linked dominant or an autosomal recessive pattern. X-linked patients have the youngest and most severe presentation and they have mutations in gp91phox, which is specific to the neutrophils. Autosomal dominant patients have mutations in one of the other 4 proteins which compose the NADPH oxidase, p47phox, p67phox, p22phox, or rarely p40phox. The immune evaluation will typically be normal other than a low neutrophil oxidative burst test, so a high level of suspicion is needed for diagnosis.
Patients with congenital neutropenia have also been found to have high IgG (Lo et al. 2013). Congenital neutropenia can be caused by multiple different genetic defects including elastase mutations (Horwitz et al. 2013). These patients have no characteristic clinical features other than neutropenia and infections. Many conditions cause congenital neutropenia (Boztug and Klein 2013) and those that have been described with hypergammaglobulinemia elsewhere in this review include ALPS, Chediak–Higashi syndrome, and Cartilage–hair hypoplasia. Many causes of congenital neutropenia include specific physical characteristics or associated features that can assist in narrowing down the diagnostic possibilities.
Hypergammaglobulinemia in predominantly antibody immunodeficiencies
Humoral immunodeficiencies are typically associated with hypogammaglobulinemia. However, there are conditions in which the immunoglobulin levels are normal (or high), but they are incapable of mounting a robust specific antibody response against infections. The International PID classification uses the term “Specific antibody deficiency with normal Ig concentrations and normal numbers of B cells” (Al-Herz et al. 2014) to refer to individuals who have normal immunoglobulins but have recurrent infections and poor responses to vaccines. This terminology does not explicitly describe that some patients with this phenotype can have high, rather than normal, immunoglobulins. Antibody deficiency with normal or elevated immunologlobulins (Nagumo and Komiyama 2000) recognizes that the immunoglobulins can be high. Some clinical immunologists may also use the term “dysgammaglobulinemia” to refer to these patients. The presence of recurrent infections with high immunoglobulins, but with a poor specific antibody production, is a phenotypic description and represents a compilation of different genetic disorders that will likely be further elucidated in the coming years. Specific antibody deficiency, otherwise known as selective IgG deficiency (Orange et al. 2012), is another phenotypic condition in which immunoglobulin levels are normal, but there is a poor response to polysaccharide antigens. This description does not currently specify that the IgG can be high. These conditions can exist on their own or as part of a broader phenotype.
Hypergammaglobulinemia in combined T-cell and B-cell immunodeficiencies
A combined immunodeficiency is one in which there is both a T-lymphocyte and B-lymphocyte deficiency.
Many different monogenic combined immunodeficiencies affecting different areas of immune system development have been reported to have elevated IgG. For example, defects in T-cell receptor development, intracellular signaling, regulatory T-cell development, endocytosis, and DNA repair have all been found to cause combined immunodeficiencies as well as high IgG.
T cells recognize antigen through their T-cell receptors. These receptors need to be created from rearranging genomic DNA. Recombinant activating gene (RAG) deficiency leads to an impaired ability to create T-cell receptors. Classically, patients with deficiencies in RAG present with Omenn's syndrome or SCID. However, patients with RAG1 deficiency can have a very variable phenotype. These patients can have prominent autoimmune features and present like patients with granulomatosis with polyangitis (Avila et al. 2010) or they can have multiple autoimmune cytopenias with IgG above the typical reference range for age (Henderson et al. 2013; Chen et al. 2014).
Lymphocytes need to transmit signals from their various cell surface receptors to the nucleus to respond to stimuli. Alterations in lymphocyte signal transduction therefore cause immunodeficiencies, including fatal immunodeficiencies. After the T-cell receptor has bound an antigen, ZAP-70 (Zeta-chain-associated protein kinase 70) is recruited to the CD3 zeta chain and plays a critical role in further downstream signaling. Mutations in ZAP-70 have been described in about 20 families and lead to a severe combined immunodeficiency syndrome characterized by recurrent candidal and viral infections as well as opportunistic infections like Pneumocystis jiroveci pneumonia (PJP). The immune profile reveals low numbers of CD8 cells, but they can have normal or elevated B cells and IgG with poor specific antibody production (Fischer et al. 2010). These patients require hematopoetic stem cell transplantation for survival.
A newly described immunodeficiency, MST1 deficiency (also known as STK4 deficiency), is characterized clinically by mild cardiac abnormalities and recurrent bacterial, candidal, and viral infections including Epstein–Barr virus (EBV) and human papillomavirus. They also have a predisposition to lymphoma and autoimmunity. Despite their hypergammaglobulinemia, they have poor B- and T-cell responses (Abdollahpour et al. 2012). MST1 appears to have a role in regulating regulatory T cells through expression of FOXP3 (Du et al. 2014) as well as regulating T-cell apoptosis and survival (Nehme et al. 2012).
Recently, mutations in LPS-responsive beige-like anchor (LRBA) have been described in patients with a variable phenotype of immune dysregulation including inflammatory bowel disease and cytopenias, immunodeficiency, or a combination of all of these features. They have been reported with low, normal, and high IgG, even with the same familial mutation (Alangari et al. 2012). The exact role of LRBA in the immune system remains to be elucidated but is thought to be involved in endocytosis.
CD27 is a cell marker for memory lymphocytes and is important for NK-cell and T-cell cytotoxicity. CD27 deficiency patients can have a widely variable phenotype within the same family. The patient in the index case initially had elevated immunoglobulins with EBV infection, he then became hypogammaglobulinemic over time and had a good result with IVIG replacement (van Montfrans et al. 2012). However, his brother died from EBV-related aplastic anemia. Overall, 10 patients have been reported and their clinical phenotype has varied from B memory cell impairment to severe problems such as chronic EBV infection or malignancy (Parvaneh et al. 2013).
Hypergammaglobulinemia in well-defined syndromes with immunodeficiency
Some immunodeficiencies have strongly associated phenotypic features such as associated neurological impairments, skeletal abnormalities, or other characteristics that can assist in the diagnosis.
Ataxia–telangestasia is an immunodeficiency due to a defect in DNA repair. These patients develop progressive cerebellar defects and have a marked predisposition to malignancy and autoimmunity. Their immune defect is variable and can include humoral immune defects. A large series of patients was described in Nowak-Wegrzyn et al. (2004) and 13% of the patients had high IgG.
Wiskott–Aldrich Syndrome (WAS) patients are well described as having elevated immunoglobulins, especially IgA and IgE, but also high IgG (Massaad et al. 2013). The WAS gene encodes for the WAS protein that transmits signals from the surface of the cell to the actin cytoskeleton. The absence of WAS protein leads to a severe multisystem disorder that includes thrombocytopenia with small platelets, eczema, recurrent infections, and a marked predisposition to develop autoimmunity and malignancy. Many of these patients require bone-marrow transplantation owing to the severity of this condition.
Patients with hyper-IgE syndrome have been described to have high IgG in addition to the more recognized high IgE. In a series of 60 patients, from France, with signal transducer and activator of transcription 3 (STAT-3) mutations from 47 kindred it was found that 27% had high IgG (Chandesris et al. 2012). This autosomal dominant form of hyper-IgE syndrome is caused by dominant negative mutations in the intracellular signaling molecule STAT-3 (Sowerwine et al. 2012). It is characterized by recurrent skin and lung infections, skeletal abnormalities such as a propensity to fractures, a high arched palate, and retained primary teeth. Hyper-IgE syndromes can also present with chronic mucocutaneous candidiasis. They have a syndrome of both inefficient infection clearance and disordered inflammation. Patients with STAT-3 mutations have laboratory findings that include increased IgE and eosinophilia and impaired intracellular signaling to a very wide variety of stimuli including the IL-6 pathway and a deficiency of IL-17A and IL-22 producing T cells as well as a deficiency of memory B cells (Chandesris et al. 2012). Some of these patients require IVIG replacement for their poor specific antibody production.
The autosomal recessive form of hyper-IgE syndrome is caused by mutations in the dedicator of cytokinesis 8 (DOCK8) (Engelhardt et al. 2009; Zhang et al. 2009). In the initial description of this condition, 6 of 11 patients had elevated IgG (Zhang et al. 2009). These patients do not have the characteristic skeletal abnormalities of autosomal dominant hyper-IgE syndrome. They have a rash similar to atopic dermatitis, atopy, recurrent sinopulmonary infections, fungal infections including chronic mucocutaneous candidiasis (CMCC), and markedly increased viral susceptibility. Severe Herpes simplex, human papilloma virus infections, and mollescum contagiosum have been described. These patients can develop malignancies related to cutaneous viral infection as well as lymphoid malignancies. Commonly, these patients have lymphopenia and eosinophilia and variable specific antibody production. Patients with DOCK8 deficiency are far more likely than patients with atopic dermatitis to have low CD4+, low CD8+, and low memory CD8+ cells (Janssen et al. 2014). DOCK8 has multiple functions in the immune system including effects on NK- and B-cell function (Nishikimi et al. 2013). Recently, an impaired ability to affinity switch immunoglobulin and make long-lived immunoglobulin responses to antigen has been demonstrated in mice with defects in this gene (Randall et al. 2009). Reduced TH-17 cells have also been described. The recognition of DOCK8 deficiency is essential because this condition can be cured with hematopoetic stem cell transplantation (Su et al. 2011).
Netherton's syndrome occurs owing to mutations in the gene SPINK5, expressed in epithelial tissue. This mutation alters the LEKT1 protein and allows for unopposed serine protease activity in the skin and results in a condition of severe congenital ichythosis and recurrent infections, especially staphylococcal and viral. As these children grow older they develop atopy. Their immune evaluations typically demonstrate low NK cells, normal or high IgG, and elevated IgE (Judge et al. 1994).
Children with mutations in ORAI1 (Feske et al. 2010) have a syndrome of hypotonia, ectodermal dysplasia, and an immunodeficiency resulting in recurrent severe infections including to opportunistic infections. Their symptoms arise owing to a defect in specialized Ca++ channel signaling, which has profound effects in the development of multiple systems. The children have been described to have normal lymphocyte numbers but poor activation and normal or elevated IgG.
Another monoallelic immunodeficiency with a recognizable phenotype has been described in patients with STAT-5b mutations. These patients have dysmorphisms, eczema, diarrhea, pneumonias, and severe herpes virus infections. Their failure to thrive is likely mutifactorial, but growth hormone signaling is significantly affected in this condition; therefore, these patients can be identified by low IGF-1, low IGFBP-3, and high prolactin. Their immune evaluation finds normal T-, B-, and NK-cell counts, and they usually have hypergammaglobulinemia and elevated CD45RO counts (Verbsky and Chatila 2013).
Mutations in the ribonuclease mitochondrial RNA processing endoribonuclease gene result in a disturbance of mitochondrial RNA processing and cell cycle control and cause a highly variable phenotype that can include a combined immunodeficiency as well as short-limbed dwarfism called Cartilage–hair hypoplasia. In a series of 12 patients, the clinical heterogeneity, even within kindred, was stressed and 1 patient was described to have IgG (18.2 g/L) at the age of 2.2 years (Kavadas et al. 2008)
Hypergammaglobulinemia in diseases of immune dysregulation
There is an ever growing appreciation that autoimmunity is a manifestation of PID. A detailed review of autoimmunity and PID is beyond the scope of this review and has been extensively reviewed recently (Goyal et al. 2009; Schuetz et al. 2010; Cunningham-Rundles 2011; Atkinson 2012; Torgerson 2012; Todoric et al. 2013). One of the described autoimmunity conditions associated with PID is autoimmune cytopenias, but many other affected organ systems have been described.
Autoimmune cytopenias are a commonly presenting sign of humoral immunodeficiency. As an example, common variable immunodeficiency (CVID) is well described as presenting with immune-mediated thrombocytopenia (Michel et al. 2004). Persistent or difficult to control autoimmune cytopenias should prompt consideration for PID. By definition, CVID does not have hypergammaglobulinemia, but other immunodeficiencies that have been described with elevated immunoglobulins can have autoimmune cytopenias. One example is DOCK8 deficiency (Zhang et al. 2009).
As a cause of autoimmune cytopenias, ALPS deserves special mention. Some patients with an ALPS-like phenotype of lymphoproliferation and hypergammaglobulinemia can have B-cell subsets similar to those in (CVID) (Rensing-Ehl et al. 2010). There is increasing recognition of an overlap of these 2 clinical presentations. Patients with ALPS typically have high IgG, and in fact it is 1 component of the revised criteria (Oliveira et al. 2010).
A mutation in the autoimmune regulatory element (AIRE) causes derangements in central T-cell tolerance (Gallo et al. 2013). When T cells are maturing, they need to learn self from non-self. The AIRE gene regulates the expression of self-antigens in the thymus so that developing T cells have exposure to self-antigens and autoreactive T cells can be deleted. Without this gene, multiple autoimmunity conditions arise. The development of autoantibodies interfering with TH-17 signaling is thought to underlie the susceptibility to candida in this condition (Puel et al. 2010a). These patients characteristically develop autoimmune polyendocrinopathy with candidiasis and ectodermal dysplasia (APECED). Patients with this condition have been reported to have elevated IgG. A large study of Finnish patients described a subset of 13 patients with skin rashes and of those 5 patients had hypergammaglobulinemia ranging from 24–51 g/L (Perheentupa 2006).
Mutations in genes essential for regulatory T-cell function can cause combined immunodeficiencies with a predisposition to marked autoimmunity. The CD25 and FOXP3 genes both encode for key proteins involved in T-regulatory cell function. Autosomal recessive CD25 deficiency was reported in humans in 1997 as a syndrome in which there is marked infiltration of T cells in the lung, liver, gut, soft tissue, and bone as well as viral, fungal, and bacterial infections. T-cell proliferation in vitro was abnormal but IgG levels were slightly increased (Sharfe et al. 1997). Mutations in FOXP3 on the X-chromosome result in a severe disruption of regulatory T cells and result in a syndrome of immunodysregulation, polyendocrinopathy, and enteropathy (IPEX). These patients often have hypergammaglobulinemia (Verbsky and Chatila 2013). Both of these conditions can be treated with hematopoietic stem cell transplantation.
The X-linked immunodeficiency XIAP deficiency causes a combined immunodeficiency in which the hallmark is susceptibility to the development of hemophaocytic lymphohistiocytosis upon exposure to EBV. In a recent report of a series of 30 patients, 1 patient was described with elevated IgG (Pachlopnik Schmid et al. 2011). The molecular defect in XIAP confers an increased susceptibility to apoptosis and it interferes with many immunological signaling pathways. Patients with XIAP deficiency are at risk of early death, so this is another very significant immunodeficiency in which the laboratory investigations can show elevated IgG.
Another immunodeficiency that has been described with hypergammaglobulinemia is Chediak--Higashi Syndrome (CHS). In a Venezuelan series, 3 out of 4 of the patients had elevated IgG (Merino et al. 1983). CHS is caused by mutations in LYST and results in abnormal organelle trafficking. These patients have a recognizable syndrome of disordered immunity in which they typically have partial occulocutaneous albinism and recurrent infections especially to Staphylococcus aureus, Streptococcus pyogenes, and Pneumococcus species. If they survive childhood they can develop significant neurological defects such as peripheral neuropathy. Many patients go on to an accelerated phase in which they develop hemophaocytic lymphohistiocytosis (Kaplan et al. 2008). Bone-marrow transplantation can cure the hematological and immunological aspects of this condition but not the neurological manifestations (Tardieu et al. 2005).
Hypergammaglobulinemia in innate immune defects
The innate immune system is so described because it consists of the first line of defense against infections without requiring prior exposure. The innate immune system recognizes preserved antigens in bacteria and viruses. Mutations in IRAK-4 affect signaling through multiple Toll-like receptors and interleukin 1 receptors (von Bernuth et al. 2012). Recently, mutations in IRAK-4 have also been demonstrated to impair T-cell activation, so there may be a larger role for IRAK-4 in adaptive immunity than currently recognized (McDonald et al. 2010). A mutation in IRAK-4 was reported to cause hypergammaglobulinemia (Ku et al. 2007) in a patient with recurrent invasive pneumococcal disease including an episode of meningitis. This patient had good vaccine responses to protein antigens but impaired responses to pneumococcal antigens and low titres to isohemagluttinins. In addition to an infectious history of severe noninvasive and invasive infections from Streptococcal pneumonia, Pseudomonas aeriginosa, and Staph aureus starting at a young age, patients with IRAK-4 deficiency tend to have a blunted inflammatory response to infections, such as the lack of a high fever in the setting of meningitis. In a large series of 48 patients with IRAK-4 deficiency, 12 patients were found to have high IgG (Picard et al. 2010).
The clinical phenotype of IRAK-4 deficiency is indistinguishable from the phenotype of another innate immune defect, MyD88 deficiency. MyD88 is upstream of IRAK-4 in the Toll-like receptor signalling pathway. Indeed, these patients can also have hypergammaglobulinemia: 4 of 12 patients with MyD88 deficiency had high IgG levels (Picard et al. 2010). This combined series of IRAK-4 and MyD88 deficient patients demonstrated the severity of these conditions with a mortality of 38%.
Downstream of MyD88 and IRAK-4 is the nuclear factor-kappa B essential modulator (NEMO). Hypomorphic mutations in NEMO can cause high IgG. In a detailed report of 72 patients (Hanson et al. 2008), 9 patients were known to have high IgG and another was listed as having high IgG1 and IgG4, so he too likely had total elevated IgG. This series reported that 2 of 10 patients died in childhood. Children with NEMO mutations have a complex phenotype. Some, but not all children, have ectodermal dysplasia and they have variable immune defects. Additionally some children have a susceptibility to mycobacteria and they may have antibody deficiency or a hyper-IgM syndrome.
Some mutations in the transcription factor, STAT-1, alter signaling of cytokines including that of IFN-gamma, IL-6, and IL-21 reduce IL-17 producing T cells and can cause CMCC, recurrent viral infections, enteropathy, mycobacterial infections, and multiple endocrine autoimmunity (Uzel et al. 2013). These patients can have elevated immunoglobulins with poor antibody production among other immune abnormalities such as lymphopenia. Importantly, high IgG is not reassuring at all in STAT1 mutations. A child with a heterozygous mutation in the DNA binding domain of STAT1 was reported to have high IgG, CMCC, and a combined, progressive immunodeficiency so profound that death occurred in childhood (Sharfe et al. 2014).
Another cause of CMCC causing hypergammaglobulinemia has been described. A mutation in IL-17RA was identified in a boy with C. albicans and S. aureus dermatitis born to consanguineous parents. This mutation caused deficiency of IL-17RA and completely abolished responses to IL-17A and IL-17F (Puel et al. 2011).
Hypergammaglobulinemia in auto-inflammatory disorders
Increasing understanding of the inflammasome and defects in its regulation has shed light on the autoinflammatory conditions. Autoinflammatory conditions include the classic periodic fever syndromes, as well as some newer conditions described such as DIRA (deficiency of the IL-1 receptor antagonist). Many autoinflammatory conditions have been reported to have hypergammaglobulinemia (Kastner et al. 2010). These conditions are well known to have high IgG and not classic primary immunodeficiencies, so they will not be discussed in detail. Tumor necrosis factor receptor-associated periodic syndrome (Cantarini et al. 2012), mutations in mevalonate kinase (Bader-Meunier et al. 2011), proteasome associated disorders (Goldbach-Mansky 2012), and Blau syndrome (Sfriso et al. 2012) have all been described with hypergammaglobulinemia.
Schnitzler syndrome is a considered an autoinflammatory syndrome with a constellation of recurrent fevers, urticarial-like rash, joint inflammation, hepatosplenomegaly, and lymphadenopathy. Although these patients typically have an elevated monoclonal IgM, there are IgG variants (Schuster et al. 2009).
Hypergammaglobulinemia in complement deficiencies
Complement is a collection of more than 40 proteins that have an important role in the innate immune system with a wide range of functions including opsonization and clearance of immune complexes (Mayilyan 2012). Patients with inherited deficiencies in complement can present with infections, autoimmunity, paroxysmal nocturnal hemoglobinuria, or uncontrolled swelling. Complement deficiency is well reported as associated with systemic lupus erythematosus (SLE) (Leffler et al. 2014). The first report of C2 deficiency with SLE and nephritis described significant hypergammaglobulinemia in this patient (Roberts et al. 1978), thus illustrating another category of immunodeficiencies that can have high IgG.
Conditions that may represent a primary immunodeficiency
Figure 1 summarizes broad conditions and presentations as discussed in this review that may be associated with both a primary immunodeficiency and hypergammaglobulinemia.
Figure 1:
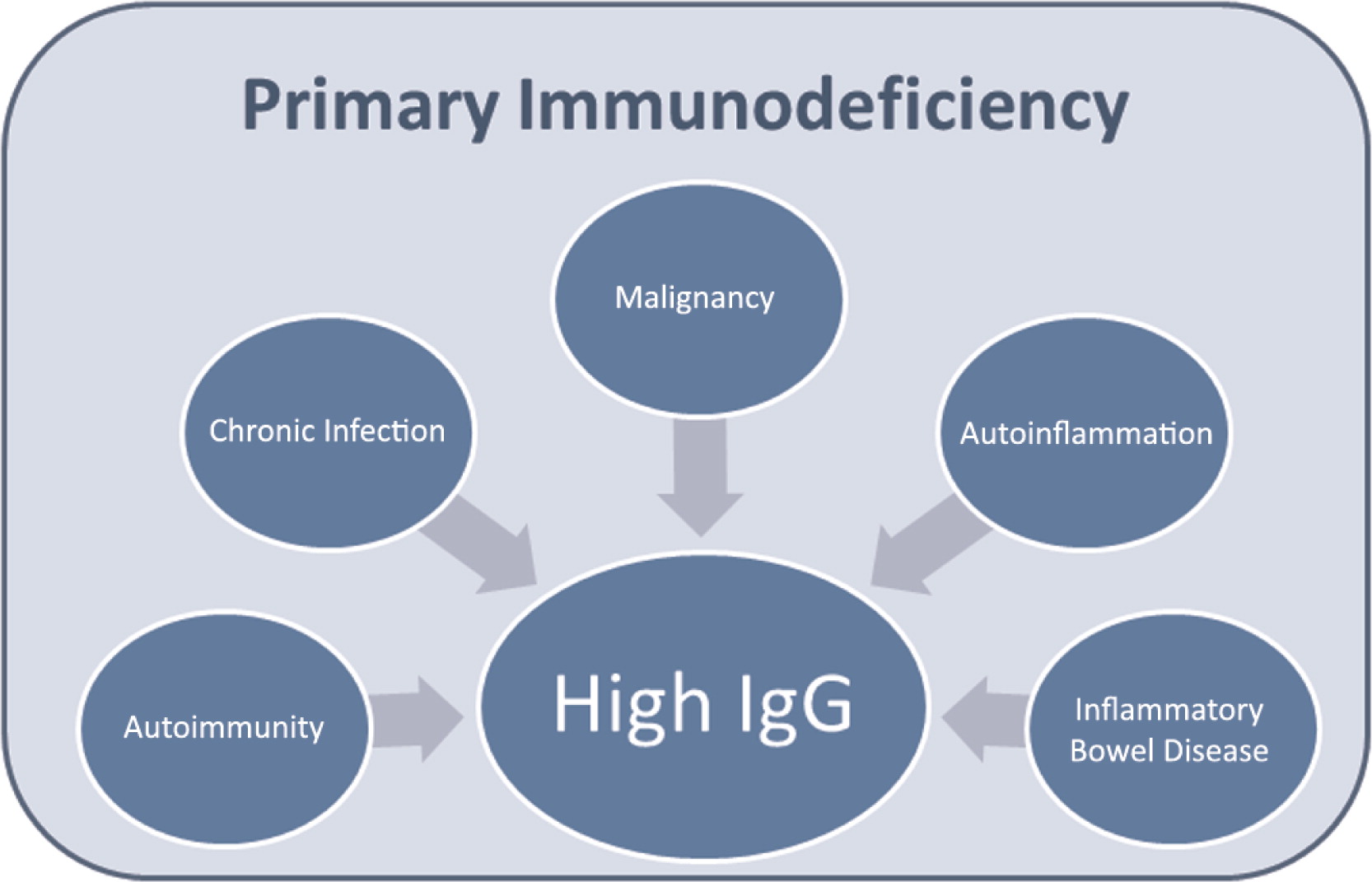
Chronic infections
Hypergammaglobulinemia is described in the setting of chronic infections (Dispenzieri et al. 2001; Lo et al. 2013). Chronic infections can of course be a major presentation of primary immunodeficiency. A few key infections will be expanded upon that demonstrate the link between chronic infection and underlying immunodeficiencies.
As discussed in earlier sections, chronic mucocutaneous candidiasis, chronic sinopulmonary infections as seen in humoral immunodeficiencies and combined immunodeficiencies, abscesses as seen in neutrophil defects, and other severe, persistent, unusual and recurrent infections can be a hallmark of primary immunodeficiency.
EBV infections are described as causing hypergammaglobulinemia (Lo et al. 2013). As discussed above, a susceptibility to herpes viruses can be due to many immunodeficiencies. Many monogenic immunodeficiencies can lead to difficulty clearing EBV including ones mentioned earlier in this review such as STK4 deficiency, XIAP, CD27 deficiency, as well as others (Parvaneh et al. 2013).
Inflammatory bowel disease
Inflammatory bowel disease (IBD) is associated with hypergammaglobulinemia (Dispenzieri et al. 2001; Lo et al. 2013). Multiple monogenic diseases involving the immune system have now been discovered that can lead to immunodysregulation and the clinical presentation of colitis (Uhlig 2013). In this review, some of these genetic causes have been discussed as combined immunodeficiencies such as LRBA deficiency, IPEX syndrome, and CD25 deficiency. Clearly, IBD can be a manifestation of an immune system disorder, so attributing the hypergammaglobulinemia to the presence of IBD may miss that the IBD and the hypergammaglobulinemia are actually symptoms of an underlying primary immunodeficiency. Although the monogenic diseases causing IBD tend to present very young, some conditions associated with chronic inflammatory GI pathology and hypergammaglobulinemia such as autosomal recessive chronic granulomatous disease and dysgammaglobulinemia can present in adults.
Malignant conditions
It is recognized in both large series of hypergammaglobulinemia (Dispenzieri et al. 2001; Lo et al. 2013) that malignancy can cause elevated immunoglobulins. Many leukemias and lymphomas have been described with high IgG. A more detailed review of immunodeficiencies that predispose to malignancies can be found elsewhere (de Miranda et al. 2011; Leechawengwongs and Shearer 2012). Multiple myeloma (Fonseca and Monge 2013) is of course 1 of the characteristic conditions that can present with elevated immunoglobulins but will not be discussed in any detail in this review.
Many of the primary immunodeficiencies previously listed in this review are strongly associated with the development of lymphoma including ataxia–telangectasia (de Miranda et al. 2011), ALPS (Rao and Oliveira 2011), and STAT-3 deficiency (Leonard et al. 2004). The recognition of single gene defects in the immune system contributing to malignancy predisposition will no doubt be further developed in the future.
Other conditions
High IgG has been noted in some benign hematological conditions such as the histiocytic disease Rosai--Dorfman (Kutlubay et al. 2014) and idiopathic plasmacytic lymphadenopathy with polyclonal hypergammaglobulinemia (Kurosawa et al. 2009) and Castleman's disease (Hanada et al. 2012). Given that the underlying genetic etiology of these conditions is not understood and there is profound immune dysregulation, they have been included in this review as conditions possibly reflective of a primary immunodeficiency.
Mechanisms of hypergammaglobulinemia
This review has documented a very wide range of primary immunodeficiency conditions that can have hypergammaglobulinemia as a feature. The mechanisms underlying the development of high IgG in these conditions is varied. Overall the etiology of hypergammaglobulinemia is not well known. Some of the proposed mechanisms will be discussed.
High IgG appears to be a non-specific reactive inflammatory marker with variable correspondence to other inflammatory makers. For example, previous work in autoimmunity showed that patients with mixed connective tissue disease had the highest IgG but only slightly increased C-reactive protein (CRP), whereas rheumatoid arthritis patients had high CRP levels and slightly increased IgG levels, and patients with SLE showed high IgG levels and low CRP levels (Bakri Hassan et al. 1998).
Hypergammaglobulinemia can also be induced in murine models of autoimmunity. In a recent report, double transgenic mice were created in which the B cells expressed a specific lambda 2 light chain and the T cells expressed a cognate T-cell receptor. These idiotype-specific T cells were found to drive oligoclonal expansion of autoreactive B cells and within a few weeks many of the B cells produced antibodies specific for anti-ds DNA and hypergammaglobulinemia (Aas-Hanssen et al. 2014).
Chronic infections may be a cause of hypergammaglobulinemia. Murine studies have shown that viruses that lead to a persistent state can lead to hypergammaglobulinemia, whereas when viruses are cleared quickly they lead to only a transient increase in IgG. The development of hypergammaglobulinemia in this model relied on cognate T-cell help, presumably for the B cell switch to IgG (Hunziker et al. 2003). Therefore, in PID, the inability to mount an effective immune response may be a cause of elevated IgG. Some infections, such as HIV, are chronic because they can evade even healthy immune systems. But in other chronic infections, consideration should be given to the host factors, such as PID, that lead to the inability to clear the infection. Certainly this chronic infection hypothesis is a likely reason for elevated IgG in patients who suffer from severe, persistent infections, but it may not be the only mechanism.
Aberrations in cytokine secretion may also contribute to hypergammaglobulinemia. Elevated IL-21, IFN-γ, IL-6, and IL-17 have been implicated in several syndromes associated with autoimmunity-related hypergammaglobulinemia (Sweet et al. 2012). Interestingly, many of the immunodeficiencies known to be associated with elevated IgG have defects affecting these pathways. For example, in STAT-3 deficient hyper-IgE syndrome, response to IL-6 and IL-10 is impaired, leading to a reduction in TH-17 responses as measured by production of IL-17 (Minegishi and Karasuyama 2009). Many of the immunodeficiencies listed in this review can present with CMCC, which is known to be associated with defects in TH-17 function (Puel et al. 2010b).
Abnormalities in cytokine production are also found in the autoinflammatory conditions. These syndromes have a marked elevation of IL-1 and often clinically respond to the IL-1 receptor blockade (Jesus and Goldbach-Mansky 2014); however, there is little information available about the response of the elevated IgG to IL-1 blockade. In Schnizler's syndrome, it has been reported that the clinical features responded to the IL-1 blockade, but the monoclonal protein (Schuster et al. 2009) did not, even after 8 months of treatment.
Suggested approach to evaluate for and treat PID in the setting of hypergammaglobulinemia
Lo et al. (2007) suggested that in pediatrics about half of the patients found to have an IgG over 20 g/L will have autoimmunity, and the likelihood of finding autoimmunity can be further improved by finding an elevated CRP, by being female, and by examining the hemoglobin and white blood cell count.
Detailed instructions of when to suspect PID are beyond the scope of this document. Warning signs of immunodeficiency (Jeffrey Model Foundation 2013) have been suggested to capture individuals suspected of immunodeficiency. These signs were evaluated recently and sensitivity and specificity were lower than ideal (Arkwright and Gennery 2011), partly because of the new understanding of what constitutes an immunodeficiency. The classic warning signs do not capture individuals with 1 very severe infection, chronic EBV, or with immune dysregulation such as that seen in early onset autoimmunity and inflammation. Screening criteria for PID will continue to evolve as the variability of existing immunodeficiencies and new immunodeficiencies are further recognized. Lists of warning signs specific to the pediatric population (Subbarayan et al. 2011) and specific to different specialists (Costa-Carvalho et al. 2014) were recently published. Immunodeficiency Canada has suggested a list of red flags that includes features of immune-dysregulation such as infantile diarrhea as well as more classic signs of immunodeficiency (Immunodeficiency Canada 2014).
Clearly, the IgG should not be the only screen for the presence of immunodeficiency, but in someone in whom immunoglobulins were ordered due to clinical suspicion for their derangement, the goal of this review is to stress that elevated immunoglobulins still leave many viable and serious diagnostic possibilities. The function of the immune system can be screened by asking about signs and symptoms of immunodeficiency as well as examining the complete blood count and differential and the specific antibody production to vaccines and prior infections (Locke et al. 2014). Table 2 presents generalized information relevant to the work-up of high immunoglobulins based on the conditions described. Other authors have suggested that an immune evaluation for patients with conditions associated with PIDs, such as autoimmune cytopenias, should be performed (Michel et al. 2004) prior to the initiation of drugs that interfere with immune assessment, such as IVIG or immunosuppressants. Clear documentation of the inability to mount appropriate vaccine responses is helpful for both elucidating an underlying diagnosis and for gaining approvals for treatments such as IgG replacement therapy.
Table 2:
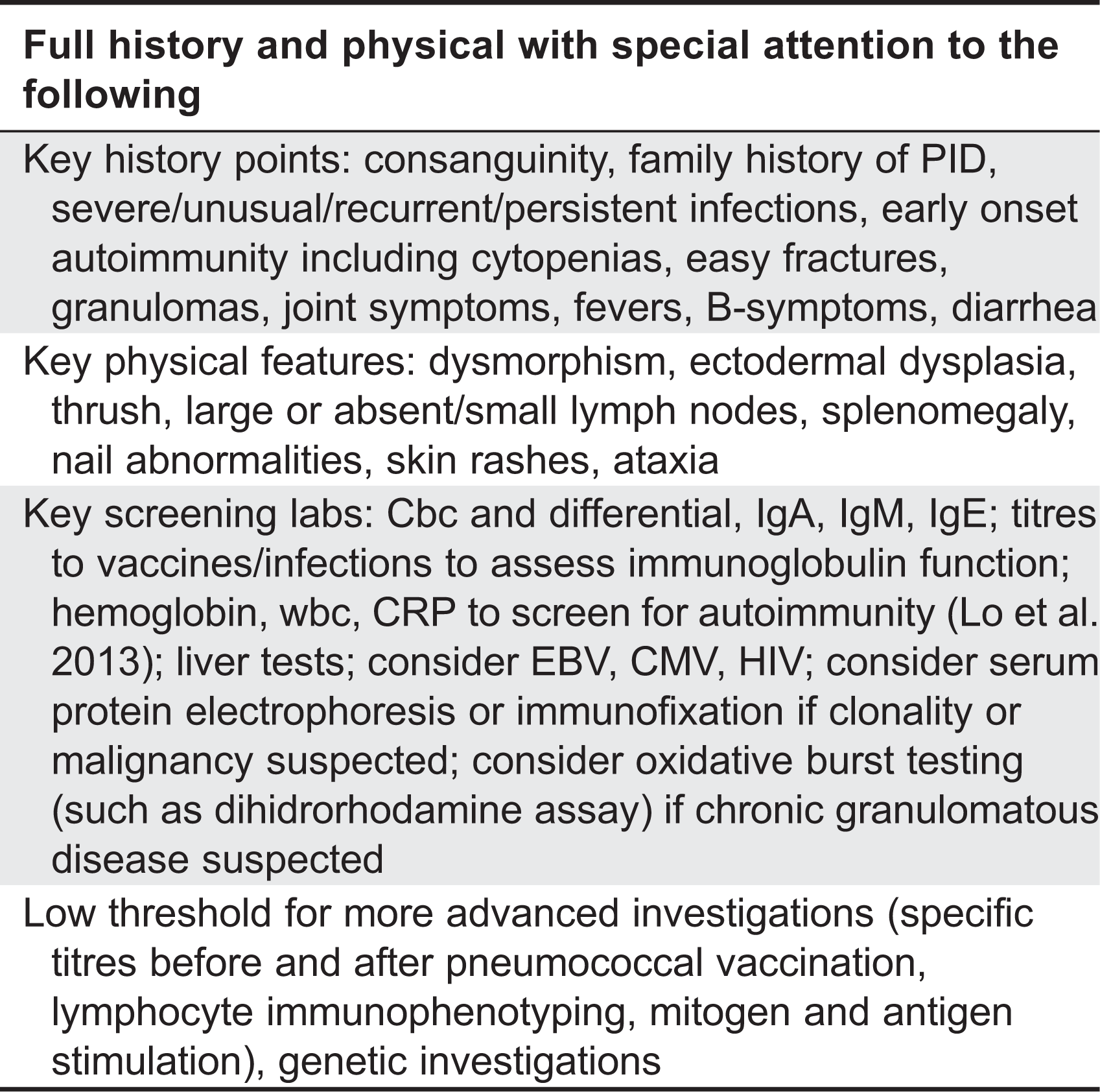
Summary
A high IgG is an uncommon finding, especially in pediatrics, and can be associated with autoimmunity, malignancy, chronic infections, inflammation, and PID, including conditions that can have fatal outcomes and those that require hematopoetic stem cell transplanation. Each of these conditions has great overlap and each can be caused by genetic abnormalities in the immune system. Defects in many aspects of the immune system including defects in neutrophils, adaptive immunity, innate immunity, and tolerance can lead to hypergammaglobulinemia. Chronic infection and abhorrent cytokine production have been proposed as mechanisms for elevated IgG.
Given the vast range of genetic disorders that can lead to hypergammaglobulinemia, it is likely that hypergammaglobulinemia can be a feature of almost any PID. Many of the immunodeficiency conditions listed here have had many patients described with high IgG. Immunologists routinely perform more detailed immune evaluations in referred patients, regardless of the IgG level. However, at the front lines of patient care there is likely less appreciation of the importance of the function of the immune system and more reliance on the numbers. An increased appreciation of elevation in IgG reflecting immune dysregulation and (or) immunodeficiency may lead to more extensive immune evaluations and earlier PID diagnoses. For example, expansion of the definition of specific antibody deficiency to explicitly include elevated IgG and the expansion of the descriptions of PIDs to include high IgG as a possible feature may prove beneficial for diagnostic awareness of immunodeficiencies.
References
Aalberse R.C., Stapel S.O., Schuurman J., and Rispens T. Immunoglobulin G4: an odd antibody Clin. Exp. Allergy. 2009 39 4 469 -477
Aas-Hanssen K., Funderud A., Thompson K.M., Bogen B., and Munthe L.A. Idiotype-specific Th cells support oligoclonal expansion of anti-dsDNA B cells in mice with lupus J. Immunol. 2014 193 6 2691 -2698
Abdollahpour H., Appaswamy G., Kotlarz D., Diestelhorst J., Beier R., Schäffer A.A., Gertz E.M., Schambach A., Kreipe H.H., Pfeifer D., Engelhardt K.R., Rezaei N., Grimbacher B., Lohrmann S., Sherkat R., and Klein C. The phenotype of human STK4 deficiency Blood. 2012 119 15 3450 -3457
Alangari A., Alsultan A., Adly N., Massaad M.J., Kiani I.S., Aljebreen A., Raddaoui E., Almomen A.-K., Al-Muhsen S., Geha R.S., and Alkuraya F.S. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency J. Allergy Clin. Immunol. 2012 130 2 481 -488.e2
Al-Herz W., Bousfiha A., Casanova J.-L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Franco J.L., Gaspar H.B., Holland S.M., Klein C., Nonoyama S., Ochs H.D., Oksenhendler E., Picard C., Puck J.M., Sullivan K., and Tang M.L.K. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency Front. Immunol. 2014 5 162
Al-Herz W. and Notarangelo L.D. Classification of primary immunodeficiency disorders: one-fits-all does not help anymore Clin. Immunol. 2012 144 1 24 -25
Arkwright P.D. and Gennery A.R. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century Ann. NY Acad. Sci. 2011 1238 7 -14
Atkinson T.P. Immune deficiency and autoimmunity Curr. Opin. Rheumatol. 2012 24 5 515 -521
Avila E.M., Uzel G., Hsu A., Milner J.D., Turner M.L., Pittaluga S., Freeman A.F., and Holland S.M. Highly variable clinical phenotypes of hypomorphic RAG1 mutations Pediatrics. 2010 126 5 e1248 -e1252
Bader-Meunier B., Florkin B., Sibilia J., Acquaviva C., Hachulla E., Grateau G., Richer O., Farber C.M., Fischbach M., Hentgen V., Jego P., Laroche C., Neven B., Lequerré T., Mathian A., Pellier I., Touitou I., Rabier D., Prieur A.-M., Cuisset L., and Quartier P. Mevalonate kinase deficiency: a survey of 50 patients Pediatrics. 2011 128 1 e152 -e159
Bakri Hassan A., Rönnelid J., Gunnarsson I., Karlsson G., Berg L., and Lundberg I. Increased serum levels of immunoglobulins, C-reactive protein, type 1 and type 2 cytokines in patients with mixed connective tissue disease J. Autoimmun. 1998 11 5 503 -508
Boztug K. and Klein C. Genetics and pathophysiology of severe congenital neutropenia syndromes unrelated to neutrophil elastase Hematol. Oncol. Clin. North Am. 2013 27 1 43 -60, vii
Cantarini L., Lucherini O.M., Muscari I., Frediani B., Galeazzi M., Brizi M.G., Simonini G., and Cimaz R. Tumour necrosis factor receptor-associated periodic syndrome (TRAPS): state of the art and future perspectives Autoimmun. Rev. 2012 12 1 38 -43
Chandesris M.-O., Melki I., Natividad A., Puel A., Fieschi C., Yun L., Thumerelle C., Oksenhendler E., Boutboul D., Thomas C., Hoarau C., Lebranchu Y., Stephan J.-L., Cazorla C., Aladjidi N., Micheau M., Tron F., Baruchel A., Barlogis V., Palenzuela G., Mathey C., Dominique S., Body G., Munzer M., Fouyssac F., Jaussaud R., Bader-Meunier B., Mahlaoui N., Blanche S., Debré M., Le Bourgeois M., Gandemer V., Lambert N., Grandin V., Ndaga S., Jacques C., Harre C., Forveille M., Alyanakian M.-A., Durandy A., Bodemer C., Suarez F., Hermine O., Lortholary O., Casanova J.-L., Fischer A., and Picard C. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey Medicine (Baltimore). 2012 91 4 e1 -e19
Chen K., Wu W., Mathew D., Zhang Y., Browne S.K., Rosen L.B., McManus M.P., Pulsipher M.A., Yandell M., Bohnsack J.F., Jorde L.B., Notarangelo L.D., and Walter J.E. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations J. Allergy Clin. Immunol. 2014 133 3 880 -882.e10
Costa-Carvalho B.T., Grumach A.S., Franco J.L., Espinosa-Rosales F.J., Leiva L.E., King A., Porras O., Bezrodnik L., Oleastro M., Sorensen R.U., and Condino-Neto A. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice J. Clin. Immunol. 2014 34 1 10 -22
Cunningham-Rundles C. Autoimmunity in primary immune deficiency: taking lessons from our patients Clin. Exp. Immunol. 2011 164 6 -11
De Miranda N.F., Björkman A., and Pan-Hammarström Q. DNA repair: the link between primary immunodeficiency and cancer Ann. NY Acad. Sci. 2011 1246 50 -63
Dispenzieri A., Gertz M.A., Therneau T.M., and Kyle R.A. Retrospective cohort study of 148 patients with polyclonal gammopathy Mayo Clin. Proc. 2001 76 5 476 -487
Du X., Shi H., Li J., Dong Y., Liang J., Ye J., Kong S., Zhang S., Zhong T., Yuan Z., Xu T., Zhuang Y., Zheng B., Geng J.-G., and Tao W. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease J. Immunol. 2014 192 4 1525 -1535
Engelhardt K.R., Mcghee S., Winkler S., Sassi A., Woellner C., Lopez-herrera G., Chen A., Kim H.S., Lloret M.G., Schulze I., Ehl S., Thiel J., Pfeifer D., Veelken H., Niehues T., Siepermann K., Weinspach S., Reisli I., Keles S., and Genel F. Large deletions and point mutations involving DOCK8 in the autosomal recessive form of the hyper-IgE syndrome J. Allergy Clin. Immunol. 2009 124 6 1289 -1302
Feske S., Picard C., and Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1 Clin. Immunol. 2010 135 2 169 -182
Fischer A., Picard C., Chemin K., Dogniaux S., le Deist F., and Hivroz C. ZAP70: a master regulator of adaptive immunity Semin. Immunopathol. 2010 32 2 107 -116
Fonseca R. and Monge J. Myeloma: classification and risk assessment Semin. Oncol. 2013 40 5 554 -566
Freeman A.F. and Holland S.M. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes Pediatr. Res. 2009 65 5 Pt 2 32R -37R
Gallo V., Giardino G., Capalbo D., Palamaro L., Romano R., Santamaria F., Maio F., Salerno M., Vajro P., and Pignata C. Alterations of the autoimmune regulator transcription factor and failure of central tolerance: APECED as a model Expert Rev. Clin. Immunol. 2013 9 1 43 -51
Geha R.S., Notarangelo L.D., Casanova J.-L., Chapel H., Conley M.E., Fischer A., Hammarström L., Nonoyama S., Ochs H.D., Puck J.M., Roifman C., Seger R., and Wedgwood J. Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee J. Allergy Clin. Immunol. 2007 120 4 776 -794
Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1 Clin. Exp. Immunol. 2012 167 3 391 -404
Goyal R., Bulua A.C., Nikolov N.P., Schwartzberg P.L., and Siegel R.M. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders Curr. Opin. Rheumatol. 2009 21 1 78 -84
Hanada T., Okuno K., Okada S., Fujimoto M., Kuranobu H., Hashida Y., Ueyama J., Murakami J., Hayashi A., Hanaki K., and Kanzaki S. Castleman disease in a child with short stature Pediatr. Int. 2012 54 5 720 -724
Hanson E.P., Monaco-Shawver L., Solt L.A., Madge L.A., Banerjee P.P., May M.J., and Orange J.S. Hypomorphic nuclear factor-kappaB essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity J. Allergy Clin. Immunol. 2008 122 6 1169 -1177.e16
Henderson L.A., Frugoni F., Hopkins G., de Boer H., Pai S.-Y., Lee Y.N., Walter J.E., Hazen M.M., and Notarangelo L.D. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity J. Allergy Clin. Immunol. 2013 132 4 969 -971.e1–2
Holland S.M. Chronic granulomatous disease Hematol. Oncol. Clin. North Am. 2013 27 1 89 -99, viii
Horwitz M.S., Corey S.J., Grimes H.L., and Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology Hematol. Oncol. Clin. North Am. 2013 27 1 19 -41, vii
Hunziker L., Recher M., Macpherson A. J., Ciurea A., Freigang S., Hengartner H., and Zinkernagel R.M. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections Nat. Immunol. 2003 4 4 343 -349
Immunodeficiency Canada. 2014. Red Flags for Primary Immunodeficiency. Retrieved September 21, 2014, from http://immunodeficiency.ca/primary-immunodeficiency/warning-signs/.
Janssen E., Tsitsikov E., Al-Herz W., Lefranc G., Megarbane A., Dasouki M., Bonilla F.A., Chatila T., Schneider L., and Geha R.S. Flow cytometry biomarkers distinguish DOCK8 deficiency from severe atopic dermatitis Clin. Immunol. 2014 150 2 220 -224
Jeffrey Model Foundation. 2013. 10 Warning Signs. Retrieved July 17, 2014, from http://www.info4pi.org/library/educational-materials/10-warning-signs.
Jesus A. A. and Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes Annu. Rev. Med. 2014 65 223 -244
Judge M.R., Morgan G., and Harper J.I. A clinical and immunological study of Netherton's syndrome Br. J. Dermatol. 1994 131 5 615 -621 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7999590
Kaplan J., De Domenico I., and Ward D.M. Chediak-Higashi syndrome Curr. Opin. Hematol. 2008 15 1 22 -29
Kastner D.L., Aksentijevich I., and Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective Cell. 2010 140 6 784 -790
Kavadas F.D., Giliani S., Gu Y., Mazzolari E., Bates A., Pegoiani E., Roifman C.M., and Notarangelo L.D. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations J. Allergy Clin. Immunol. 2008 122 6 1178 -1184
Ku C.-L., von Bernuth H., Picard C., Zhang S.-Y., Chang H.-H., Yang K., Chrabieh M., Issekutz A.C., Cunningham C.K., Gallin J., Holland S.M., Roifman C., Ehl S., Smart J., Tang M., Barrat F.J., Levy O., McDonald D., Day-Good N.K., Miller R., Takada H., Hara T., Al-Hajjar S., Al-Ghonaium A., Speert D., Sanlaville D., Li X., Geissmann F., Vivier E., Maródi L., Garty B.-Z., Chapel H., Rodriguez-Gallego C., Bossuyt X., Abel L., Puel A., and Casanova J.-L. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity J. Exp. Med. 2007 204 10 2407 -2422
Kurosawa S., Akiyama N., Ohwada A., Warabi M., Suenaga M., Kojima M., and Tomiyama J. Idiopathic plasmacytic lymphadenopathy with polyclonal hypergammaglobulinemia accompanied with cutaneous involvement and renal dysfunction Jpn. J. Clin. Oncol. 2009 39 10 682 -685
Kutlubay Z., Bairamov O., Sevim A., Demirkesen C., and Mat M.C. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature Am. J. Dermatopathol. 2014 36 4 353 -357
Leechawengwongs E. and Shearer W.T. Lymphoma complicating primary immunodeficiency syndromes Curr. Opin. Hematol. 2012 19 4 305 -312
Leffler J., Bengtsson A.A., and Blom A.M. The complement system in systemic lupus erythematosus: an update Ann. Rheum. Dis. 2014 73 9 1601 -1606
Leonard G.D., Posadas E., Herrmann P.C., Anderson V.L., Jaffe E.S., Holland S.M., and Wilson W.H. Non-Hodgkin's lymphoma in Job's syndrome: a case report and literature review Leuk. Lymphoma. 2004 45 12 2521 -2525
Lo M.S., Zurakowski D., Son M.B.F., and Sundel R.P. Hypergammaglobulinemia in the pediatric population as a marker for underlying autoimmune disease: a retrospective cohort study Pediatr. Rheumatol. Online J. 2013 11 1 42
Locke B.A., Dasu T., and Verbsky J.W. Laboratory diagnosis of primary immunodeficiencies Clin. Rev. Allergy Immunol. 2014 46 2 154 -168
Maggina P. and Gennery A.R. Classification of primary immunodeficiencies: need for a revised approach? J. Allergy Clin. Immunol. 2013 131 2 292 -294
Massaad M.J., Ramesh N., and Geha R.S. Wiskott-Aldrich syndrome: a comprehensive review Ann. NY Acad. Sci. 2013 1285 26 -43
Mayilyan K.R. Complement genetics, deficiencies, and disease associations Prot. Cell. 2012 3 7 487 -496
McDonald D.R., Goldman F., Gomez-Duarte O.D., Issekutz A.C., Kumararatne D.S., Doffinger R., and Geha R.S. Impaired T-cell receptor activation in IL-1 receptor-associated kinase-4-deficient patients J. Allergy Clin. Immunol. 2010 126 2 332 -337.e1–2
Merino F., Klein G.O., Henle W., Ramirez-Duque P., Forsgren M., and Amesty C. Elevated antibody titers to Epstein-Barr virus and low natural killer cell activity in patients with Chediak-Higashi syndrome Clin. Immunol. Immunopathol. 1983 27 3 326 -339
Michel M., Chanet V., Galicier L., Ruivard M., Levy Y., Hermine O., Oksenhendler E., Schaeffer A., Bierling P., and Godeau B. Autoimmune thrombocytopenic purpura and common variable immunodeficiency: analysis of 21 cases and review of the literature Medicine (Baltimore). 2004 83 4 254 -263 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15232313
Minegishi Y. and Karasuyama H. Defects in Jak-STAT-mediated cytokine signals cause hyper-IgE syndrome: lessons from a primary immunodeficiency Int. Immunol. 2009 21 2 105 -112
Nagumo H. and Komiyama A. [Antibody deficiency with normal(or elevated) immunoglobulins] Ryoikibetsu Shokogun Shirizu. 2000 32 66 -68 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11212825
Nehme N.T., Pachlopnik Schmid J., Debeurme F., André-Schmutz I., Lim A., Nitschke P., Rieux-Laucat F., Lutz P., Picard C., Mahlaoui N., Fischer A., and de Saint Basile G. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival Blood. 2012 119 15 3458 -3468
Nishikimi A., Kukimoto-Niino M., Yokoyama S., and Fukui Y. Immune regulatory functions of DOCK family proteins in health and disease Exp. Cell Res. 2013 319 15 2343 -2349
Notarangelo L.D., Fischer A., Geha R.S., Casanova J.-L., Chapel H., Conley M.E., Cunningham-Rundles C., Etzioni A., Hammartröm L., Nonoyama S., Ochs H.D., Puck J., Roifman C., Seger R., and Wedgwood J. Primary immunodeficiencies: 2009 update J. Allergy Clin. Immunol. 2009 124 6 1161 -1178
Nowak-Wegrzyn A., Crawford T.O., Winkelstein J.A., Carson K.A., and Lederman H.M. Immunodeficiency and infections in ataxia-telangiectasia J. Pediatr. 2004 144 4 505 -511
Ochs H.D. Patients with abnormal IgM levels: assessment, clinical interpretation, and treatment Ann. Allergy. Asthma Immunol. 2008 100 5 509 -511
Oliveira J.B., Bleesing J.J., Dianzani U., Fleisher T.A., Jaffe E.S., Lenardo M.J., Rieux-laucat F., Siegel R.M., Su H.C., Teachey D.T., and Rao V.K. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. 2010 116 14 35 -40
Orange J.S., Ballow M., Stiehm E.R., Ballas Z.K., Chinen J., De La Morena M., Kumararatne D., Harville T.O., Hesterberg P., Koleilat M., McGhee S., Perez E.E., Raasch J., Scherzer R., Schroeder H., Seroogy C., Huissoon A., Sorensen R.U., and Katial R. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the American Academy of Allergy, Asthma & Immunology J. Allergy Clin. Immunol. 2012 130 3 S1 -S24
Pachlopnik Schmid J., Canioni D., Moshous D., Touzot F., Mahlaoui N., Hauck F., Kanegane H., Lopez-Granados E., Mejstrikova E., Pellier I., Galicier L., Galambrun C., Barlogis V., Bordigoni P., Fourmaintraux A., Hamidou M., Dabadie A., Le Deist F., Haerynck F., Ouachée-Chardin M., Rohrlich P., Stephan J.-L., Lenoir C., Rigaud S., Lambert N., Milili M., Schiff C., Chapel H., Picard C., de Saint Basile G., Blanche S., Fischer A., and Latour S. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011 117 5 1522 -1529
Parvaneh N., Filipovich A.H., and Borkhardt A. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases Br. J. Haematol. 2013 162 5 573 -586
Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy J. Clin. Endocrinol. Metab. 2006 91 8 2843 -2850
Picard C., von Bernuth H., Ghandil P., Chrabieh M., Levy O., Arkwright P.D., McDonald D., Geha R.S., Takada H., Krause J.C., Creech C.B., Ku C.-L., Ehl S., Maródi L., Al-Muhsen S., Al-Hajjar S., Al-Ghonaium A., Day-Good N.K., Holland S.M., Gallin J.I., Chapel H., Speert D.P., Rodriguez-Gallego C., Colino E., Garty B.-Z., Roifman C., Hara T., Yoshikawa H., Nonoyama S., Domachowske J., Issekutz A.C., Tang M., Smart J., Zitnik S.E., Hoarau C., Kumararatne D.S., Thrasher A.J., Davies E.G., Bethune C., Sirvent N., de Ricaud D., Camcioglu Y., Vasconcelos J., Guedes M., Vitor A.B., Rodrigo C., Almazán F., Méndez M., Aróstegui J.I., Alsina L., Fortuny C., Reichenbach J., Verbsky J.W., Bossuyt X., Doffinger R., Abel L., Puel A., and Casanova J.-L. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency Medicine (Baltimore). 2010 89 6 403 -425
Puel A., Cypowyj S., Bustamante J., Wright J.F., Liu L., Lim H.K., Migaud M., Israel L., Chrabieh M., Audry M., Gumbleton M., Toulon A., Bodemer C., El-Baghdadi J., Whitters M., Paradis T., Brooks J., Collins M., Wolfman N.M., Al-Muhsen S., Galicchio M., Abel L., Picard C., and Casanova J.-L. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity Science. 2011 332 6025 65 -68
Puel A., Döffinger R., Natividad A., Chrabieh M., Barcenas-Morales G., Picard C., Cobat A., Ouachée-Chardin M., Toulon A., Bustamante J., Al-Muhsen S., Al-Owain M., Arkwright P.D., Costigan C., McConnell V., Cant A.J., Abinun M., Polak M., Bougnères P.-F., Kumararatne D., Marodi L., Nahum A., Roifman C., Blanche S., Fischer A., Bodemer C., Abel L., Lilic D., and Casanova J.-L. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I J. Exp. Med. 2010a 207 2 291 -297
Puel A., Picard C., Cypowyj S., Lilic D., Abel L., and Casanova J.-L. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr. Opin. Immunol. 2010b 22 4 467 -474
Randall K.L., Lambe T., Johnson A.L., Johnson A., Treanor B., Kucharska E., Domaschenz H., Whittle B., Tze L.E., Enders A., Crockford T.L., Bouriez-Jones T., Alston D., Cyster J.G., Lenardo M.J., Mackay F., Deenick E.K., Tangye S.G., Chan T.D., Camidge T., Brink R., Vinuesa C.G., Batista F.D., Cornall R.J., and Goodnow C.C. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production Nat. Immunol. 2009 10 12 1283 -1291
Rao V.K. and Oliveira J.B. How I treat autoimmune lymphoproliferative syndrome Blood. 2011 118 22 5741 -5751
Rensing-Ehl A., Warnatz K., Fuchs S., Schlesier M., Salzer U., Draeger R., Bondzio I., Joos Y., Janda A., Gomes M., Abinun M., Hambleton S., Cant A., Shackley F., Flood T., Waruiru C., Beutel K., Siepermann K., Dueckers G., Niehues T., Wiesel T., Schuster V., Seidel M.G., Minkov M., Sirkiä K., Kopp M.V., Korhonen M., Schwarz K., Ehl S., and Speckmann C. Clinical and immunological overlap between autoimmune lymphoproliferative syndrome and common variable immunodeficiency Clin. Immunol. 2010 137 3 357 -365
Roberts J.L., Schwartz M.M., and Lewis E.J. Hereditary C2 deficiency and systemic lupus erythematosus associated with severe glomerulonephritis Clin. Exp. Immunol. 1978 31 2 328 -338 Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1541209&tool=pmcentrez&rendertype=abstract
Schauer U., Stemberg F., Rieger C.H.L., Borte M., Schubert S., Riedel F., Herz U., Renz H., Wick M., Carr-Smith H.D., Bradwell A.R., and Herzog W. IgG subclass concentrations in certified reference material 470 and reference values for children and adults determined with the binding site reagents Clin. Chem. 2003 49 11 1924 -1929 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14578325
Schuetz C., Niehues T., Friedrich W., and Schwarz K. Autoimmunity, autoinflammation and lymphoma in combined immunodeficiency (CID) Autoimmun. Rev. 2010 9 7 477 -482
Schuster C., Kränke B., Aberer E., Arbab E., Sturm G., and Aberer W. Schnitzler syndrome: response to anakinra in two cases and a review of the literature Int. J. Dermatol. 2009 48 11 1190 -1194
Sfriso P., Caso F., Tognon S., Galozzi P., Gava A., and Punzi L. Blau syndrome, clinical and genetic aspects Autoimmun. Rev. 2012 12 1 44 -51
Sharfe N., Dadi H.K., Shahar M., and Roifman C.M. Human immune disorder arising from mutation of the chain of the interleukin-2 receptor Proc. Natl. Acad. Sci. 1997 94 7 3168 -3171
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., Herbrick J.-A., and Roifman C.M. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1 J. Allergy Clin. Immunol. 2014 133 3 807 -817
Sowerwine K.J., Holland S.M., and Freeman A.F. Hyper-IgE syndrome update Ann. NY Acad. Sci. 2012 1250 25 -32
Su H.C., Jing H., and Zhang Q. DOCK8 deficiency Ann. NY Acad. Sci. 2011 1246 26 -33
Subbarayan A., Colarusso G., Hughes S.M., Gennery A.R., Slatter M., Cant A.J., and Arkwright P.D. Clinical features that identify children with primary immunodeficiency diseases Pediatrics. 2011 127 5 810 -816
Sweet R.A., Lee S.K., and Vinuesa C.G. Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity Curr. Opin. Immunol. 2012 24 6 658 -664
Tardieu M., Lacroix C., Basile D. Saint, and Fischer A. Brief report progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak-Higashi syndrome Blood. 2005 106 1 40 -42
Todoric K., Koontz J.B., Mattox D., and Tarrant T.K. Autoimmunity in immunodeficiency Curr. Allergy Asthma Rep. 2013 13 4 361 -370
Torgerson T.R. Immunodeficiency diseases with rheumatic manifestations Pediatr. Clin. North Am. 2012 59 2 493 -507
Uhlig H.H. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease Gut. 2013 62 12 1795 -1805
Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E., Bonilla F.A, Pechacek J., Chandrasekaran P., Browne S.K., Agharahimi A., Gharib A.M., Mannurita S.C., Yim J.J., Gambineri E., Torgerson T., Tran D.Q., Milner J.D., and Holland S.M. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome J. Allergy Clin. Immunol. 2013 131 6 1611 -1623
Van Montfrans J.M., Hoepelman A.I.M., Otto S., van Gijn M., van de Corput L., de Weger R.A., Monaco-Shawver L., Banerjee P.P., Sanders E.A.M., Jol-van der Zijde C.M., Betts M.R., Orange J.S., Bloem A.C., and Tesselaar K. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia J. Allergy Clin. Immunol. 2012 129 3 787 -793.e6
Verbsky J.W. and Chatila T.A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases Curr. Opin. Pediatr. 2013 25 6 708 -714
Villa A., Notarangelo L.D., and Roifman C.M. Omenn syndrome: inflammation in leaky severe combined immunodeficiency J. Allergy Clin. Immunol. 2008 122 6 1082 -1086
Von Bernuth H., Picard C., Puel A., and Casanova J.-L. Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans Eur. J. Immunol. 2012 42 12 3126 -3135
Wood P., Stanworth S., Burton J., Jones A, Peckham D.G., Green T., Hyde C., and Chapel H. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review Clin. Exp. Immunol. 2007 149 3 410 -423
Zhang Q., Davis J.C., Lamborn I.T., Freeman A.F., Jing H., Favreau A.J., Matthews H.F., Davis J., Turner M.L., Uzel G., Holland S.M., and Su H.C. Combined immunodeficiency associated with DOCK8 mutations N. Engl. J. Med. 2009 361 21 2046 -2055
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 2 • June 2015
Pages: 57 - 73
History
Received: 12 December 2014
Accepted: 17 December 2014
Accepted manuscript online: 17 December 2014
Version of record online: 18 December 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Julia Upton. 2015. Immunodeficiencies with hypergammaglobulinemia: a review. LymphoSign Journal.
2(2): 57-73. https://doi.org/10.14785/lpsn-2014-0019
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. Elevated serum gamma globulins in apparently healthy Nigerians living in Ogbomoso: a possible manifestation of phagocytic dysfunction
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
