Atypical presentation and manifestations in X-linked agammaglobulinemia patients with novel BTK mutations
Abstract
X-linked agammaglobulinemia (XLA) is a rare immunodeficiency caused by defects in the Bruton tyrosine kinase (BTK) gene, characterized by impaired B-cell development, reduced immunoglobulin production, and increased susceptibility to bacterial infections at an early age. Some XLA patients show atypical presentations, with most reports concentrating on the diagnosis at a relatively old age. They presented with infections at late age or with unusual pathogens; however, other atypical manifestations have only rarely been reported.
Methods: Description of patients with XLA and novel mutations in BTK who presented with atypical manifestations or developed noninfectious complications.
Results: Four patients presented unique manifestations unusual for XLA. The first with Granulomatous Dermatitis, the second with acute demyelinating encephalomyelitis, the third with “Crohn's disease like” localized protein-losing enteropathy, and the last patient with idiopathic thrombocytopenic purpura, which is an unexpected finding in a patient devoid of endogenous immunoglobulins. Mutations in BTK were found in all domains of the gene; 1 resulted in a stop codon and 3 were missense mutations.
Conclusions: Early recognition of atypical presentations and manifestations of patients with XLA is crucial for timely initiation of life-saving therapy, which may include anti-bacterial and anti-inflammatory treatments in addition to immunoglobulin.
Statement of novelty: In this study we present unique inflammatory and autoimmune phenomenons in XLA patients that were not described previously and are somewhat unexpected. These should alert the immunologist for the possibility of XLA diagnosis.
Introduction
X-linked agammaglobulinemia (XLA), or Bruton's disease (Bruton 1952), is a rare immunodeficiency in which mutations in the BTK gene cause an arrest of B-cell development with a lack of peripheral mature B cells and profound hypogammaglobulinemia. As a result, patients have an increased susceptibility to a variety of encapsulated bacteria and enteroviruses; microorganisms for which antibodies play a critical role in host defense. According to a U.S. registry, the most common presentations leading to diagnosis were: infections in 85% of patients (most common), positive family history (second most common), and neutropenia (third most common) (Winkelstein et al. 2006). The most common infections are sinopulmonary: 50% of the patients present by 1 year of age and almost all of the patients present by the age of 5 years. The most common pathogens are Streptococcus pneumoniae, Haemophilus influenzae, and Pseudomonas spp. However, some patients have an atypical presentation. Because XLA is a rare disease, relatively few studies reporting atypical presentations have been published, most of them show late presentation, and very few describe atypical presentation in the young age group (Hashimoto et al. 1999; Stewart et al. 2001). Among these unusual presentations are reports of patients with pyoderma skin lesions (Lin et al. 2006), and Pneumocystis carinii pneumonitis (Saulsbury et al. 1979). Studies attempting to investigate genotype–phenotype correlation suggested that missense mutations in the BTK gene that affect nonconserved regions of the molecule are less severe (López-Granados et al. 2005) and that mutations that preserve expression of the protein usually have a higher number of B cells (Broides et al. 2006); however, there are many exceptions to these rules.
In this article we present 4 patients with unusual presentations or manifestations, some of which are unexpected in XLA patients such as autoimmune phenomenon.
Methods
Patients
We reviewed the medical records of all patients followed in our immunology clinic who were diagnosed with XLA. Research ethics board approval was attained for this study. Genetic tests were performed with informed consent from either a guardian of the patient or the patient.
Flow cytometry
Peripheral blood mononuclear cells were obtained by Ficoll–Hypaque density gradient centrifugation and surface phenotypes were determined by flow cytometry on a Coulter EPICS V flow cytometer (Coulter). All fluorescent labeled antibodies were purchased from eBiosciences (San Diego, USA).
Serum concentration of immunoglobulins
Serum concentrations of immunoglobulins were measured by nephelometry.
Genetic analysis
All exons and all exon–intron boundaries of the BTK gene were amplified using polymerase chain reaction and sequenced with an automated sequencer (Beckman Coulter CEQ8000) and compared with the normal gene sequences. Polyphen scores showed significant predictive impact of the substitution on protein structure and function.
Results
Patient 1
This patient was born after a normal pregnancy to nonconsanguineous parents and presented at 15 months of age with fever, rash, and neutropenia. Five months later he experienced another febrile illness and again blood film showed he had neutropenia. On examination he had no dysmorphic features, weight and height were at the 75th percentile, and development was appropriate for his age. He later developed recurrent pruritic erythematous papular lesions on his trunk and extremities. Biopsies revealed multiple granulomas with scattered Langerhans-type giant cells and extensive mononuclear infiltration consistent with granulomatous disease.
Bacterial, mycobacteria, and fungal stains were negative. The patient did not have a fever, and he had no increased inflammatory markers. Thorough radiologic evaluation did not reveal evidence for visceral granulomatous disease. The skin lesions slowly resolved with topical steroids and anti-bacterial treatment.
Patient 2
This patient was a 9-year-old boy who was transferred to our hospital with intractable seizures. His illness followed an upper respiratory viral disease. A week after his acute febrile illness, which was considered an upper respiratory infection, he developed ataxia, dysarthria, and dysphagia without fever. The initial sepsis workup revealed a white blood cell count of 2.6 × 103 cell/µL, and lumbar puncture showed cerebral spinal fluid (CSF) with a high protein level of 889 mg/L (100–400 normal range) but no elevation of white blood cell count, normal glucose, and no organisms in the gram stain or cultures (before initiation of any antimicrobial treatment). Imaging of his brain showed areas of demyelination that were typical of acute demyelinating encephalomyelitis.
Past medical history revealed one occasion of suspected pneumonia that was treated with a course of erythromycin, and it resolved quickly. Physical examination revealed small cervical lymph nodes, and tonsillar tissue was present.
Family history was unremarkable with nonconsanguineous parents and 3 healthy siblings. There was no history of recurrent infections or early childhood death in the family.
The patient was treated with steroids, acyclovir, dilantin, and intravenous immunoglobulins (IVIg); he slowly and fully recovered and continues on IVIg treatment.
Patient 3
This patient was a boy, 3 years and 10 months of age, who was diagnosed initially because of a recurrent, red, fine, nonitchy rash that persisted for a week involving the trunk and extremities including palms and soles. During early events he was afebrile and well, but later on these episodes were associated with fever. Serology tests for viruses, including measles, which the child was vaccinated for, were negative. During the period of investigation he developed thrombocytopenia (platelets 40 000 cells/µL) and a bone marrow aspiration showed normal cellularity with adequate numbers of megakaryocytes as well as normal erythropoiesis and granulopoiesis. Upon starting replacement with IVIg, the thrombocytopenia resolved suggesting an underlying autoimmune mechanism.
The child's past medical history revealed several episodes of acute otitis media that resolved after the insertion of tympanic ventilation tubes. He did not experience pneumonia or other invasive infections. He did not require intravenous antibiotics treatment at any time.
Family history did not reveal any individuals with recurrent infection or death in early childhood caused by infections. On physical examination, small cervical lymph nodes were palpated and tonsillar tissue could be clearly visualized.
Patient 4
This patient was a boy who presented at the age of 1 year 10 months with diarrhea lasting for 2 months. An initial investigation recovered Giardia Lamblia from the stool and treatment with Flagyl resulted in improvement. Six months later he developed fever, perianal ulcers, and bloody stools. No infectious cause was found and gastrointestinal (GI) studies revealed thickening of the bowel wall and numerous pseudopolyps.
Immune workup at this point revealed that immunoglobulins were absent. Flow cytometry showed near-absence of B lymphocytes, 0.2% of the total number.
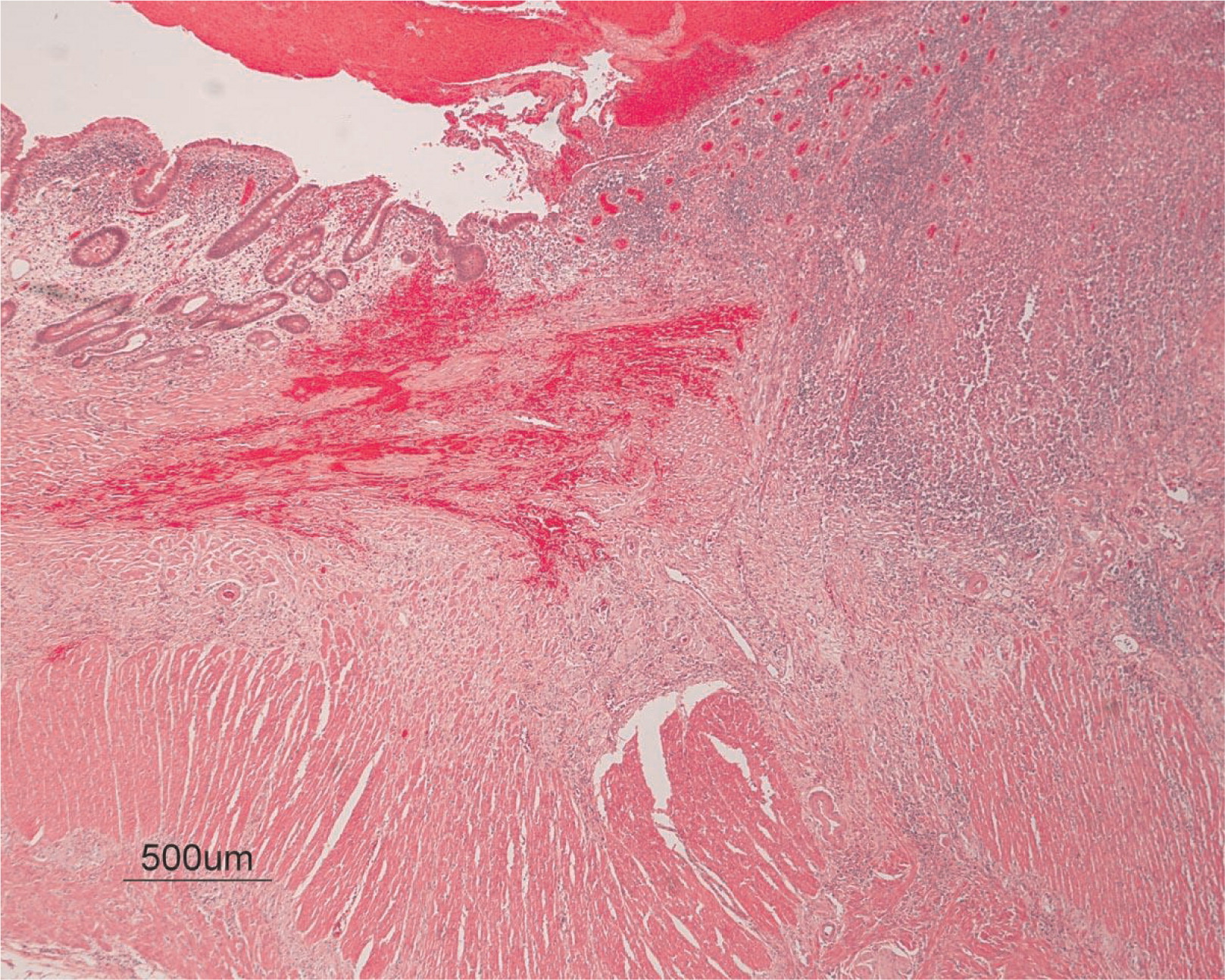
He continued to experience bouts of diarrhea, leading to failure to thrive. Further studies showed protein-losing enteropathy and repeated endoscopies with biopsies showed distortion of villous architecture with moderate crypt hyperplasia, absence of plasma cells, and decreased lymphoid cells in the lamina propria. At the age of 13 years he was admitted for left-sided abdominal pain and was noted to have lower left abdominal tenderness. Bowel contrast studies (Figures 1–3) revealed a consistent filling defect of mid-jejunum and a CT scan demonstrated focal small bowel thickening and a possible submucosal process, dubbed “Crohn's like”. Symptoms intensified and he underwent a laparotomy during which a 77 cm segment of jejunum was resected. Examination of the resected segment demonstrated numerous pseudopolyps with extensive foci of ulcerations, congestions, and hemorrhages, with total effacement of mucosal architecture (Figure 4). On microscopy, the ulcerated segment revealed acute on chronic inflammation. The acute inflammation was characterized by the presence of cryptitis (Figure 5). The lamina propria was expanded by increased numbers of mononuclear cells but no plasma cells were identified. In addition to the presence of increased infiltrates of chronic inflammatory lymphocytes, the lamina propria also showed underlying chronic regenerative changes such as glandular branching, focal reduplication of the muscularis mucosae, and hypertrophy of the muscularis propria (Figures 6 and 7). No granulomas or granulomatous inflammatory infiltrates were found. In addition, gastric metaplasia was seen in the submucosa (Figure 7). In some areas of the ulcer the muscosa was replaced by acutely inflamed granulation tissue (Figures 4 and 7). Intense infiltrates of neutrophils extended from the ulcerated lamina propria deep into the submucosa and through the muscularis propria (Figures 7 and 8). The inflammation resulted in the development of edema, mild fibrosis, and thickening of the serosa (Figure 9). All of these changes in the bowel wall correlated with the Crohn's-like radiologic changes that were detected. Despite the pan-mural ileitis, a diagnosis of Crohn's ileitis could not be made because of the absence of granuloma formations. The diagnosis of bacterial overgrowth was also not supported as all bacterial, fungal, and protozoal stains were negative.
Figure 1:
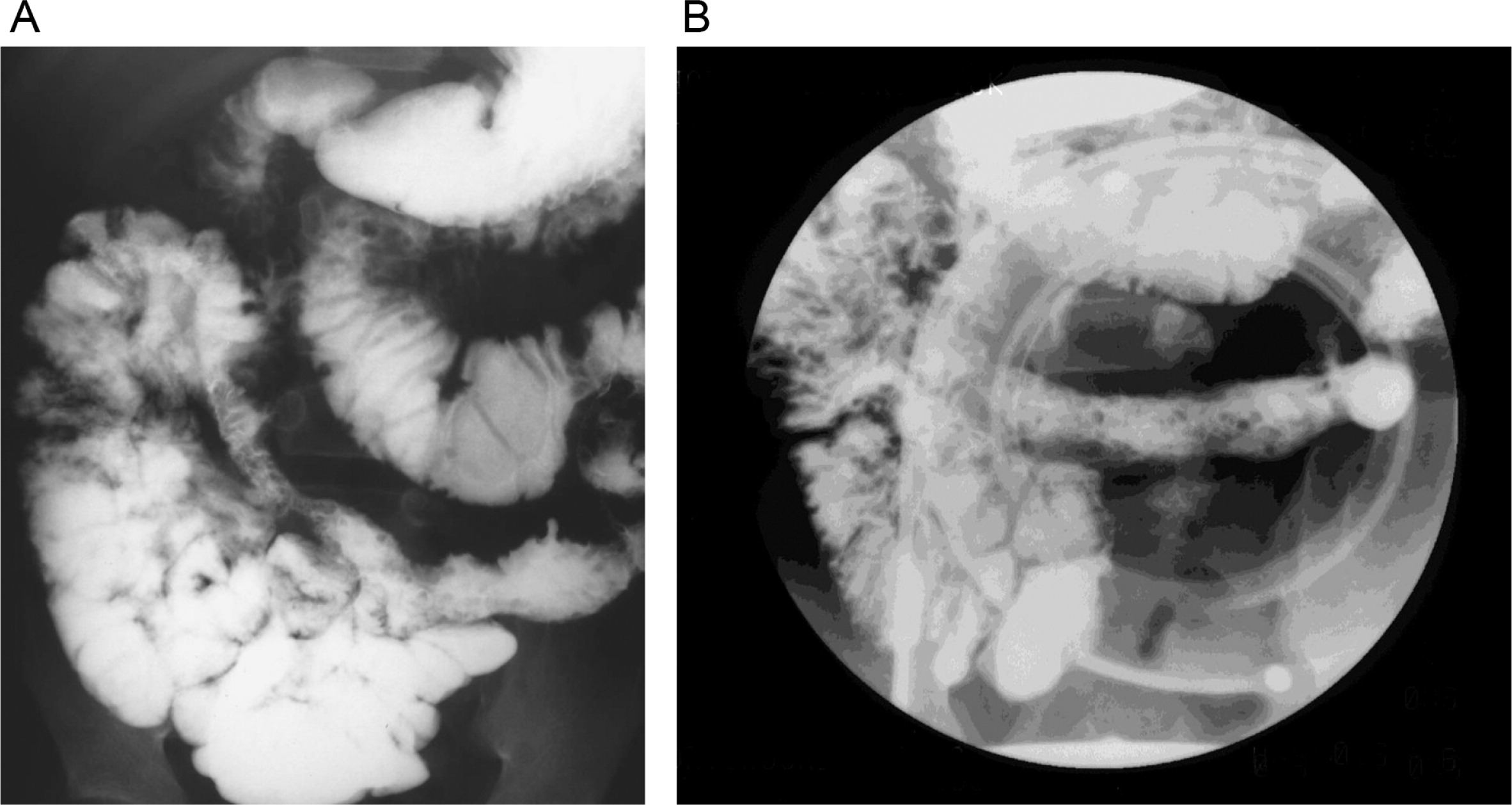
Figure 2:
Figure 3:
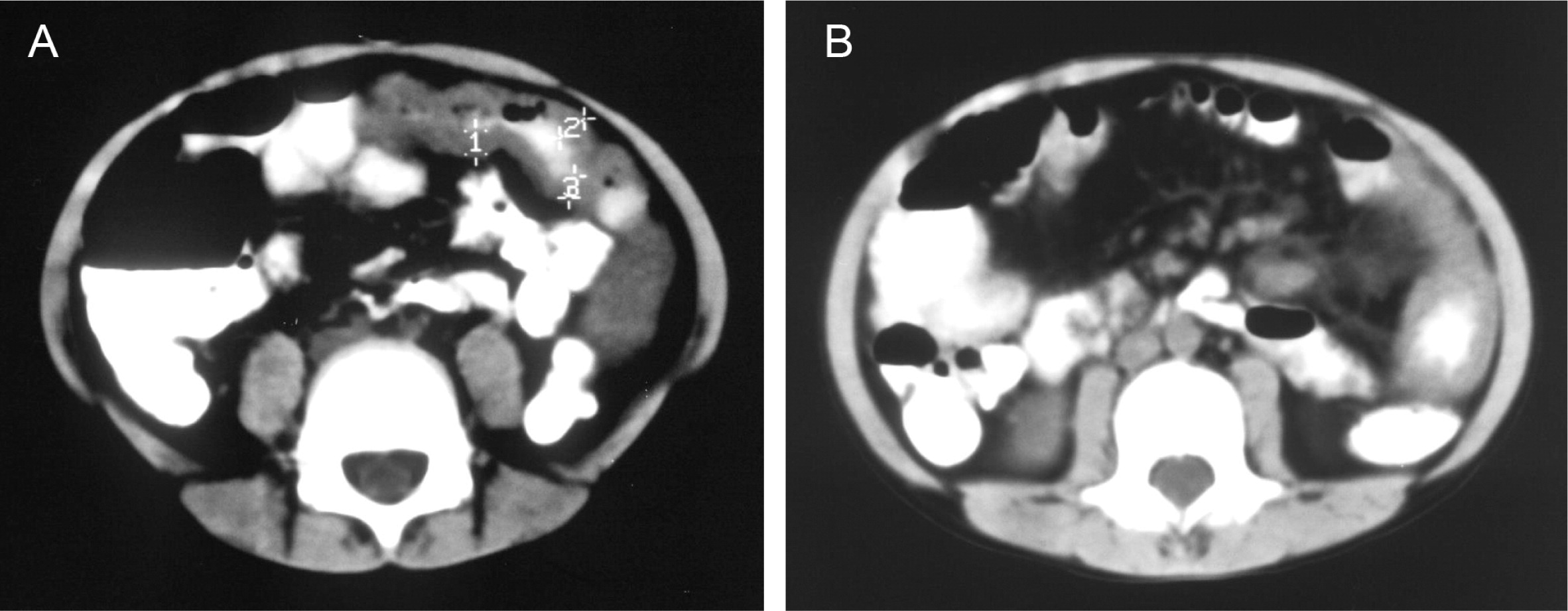
Figure 4:
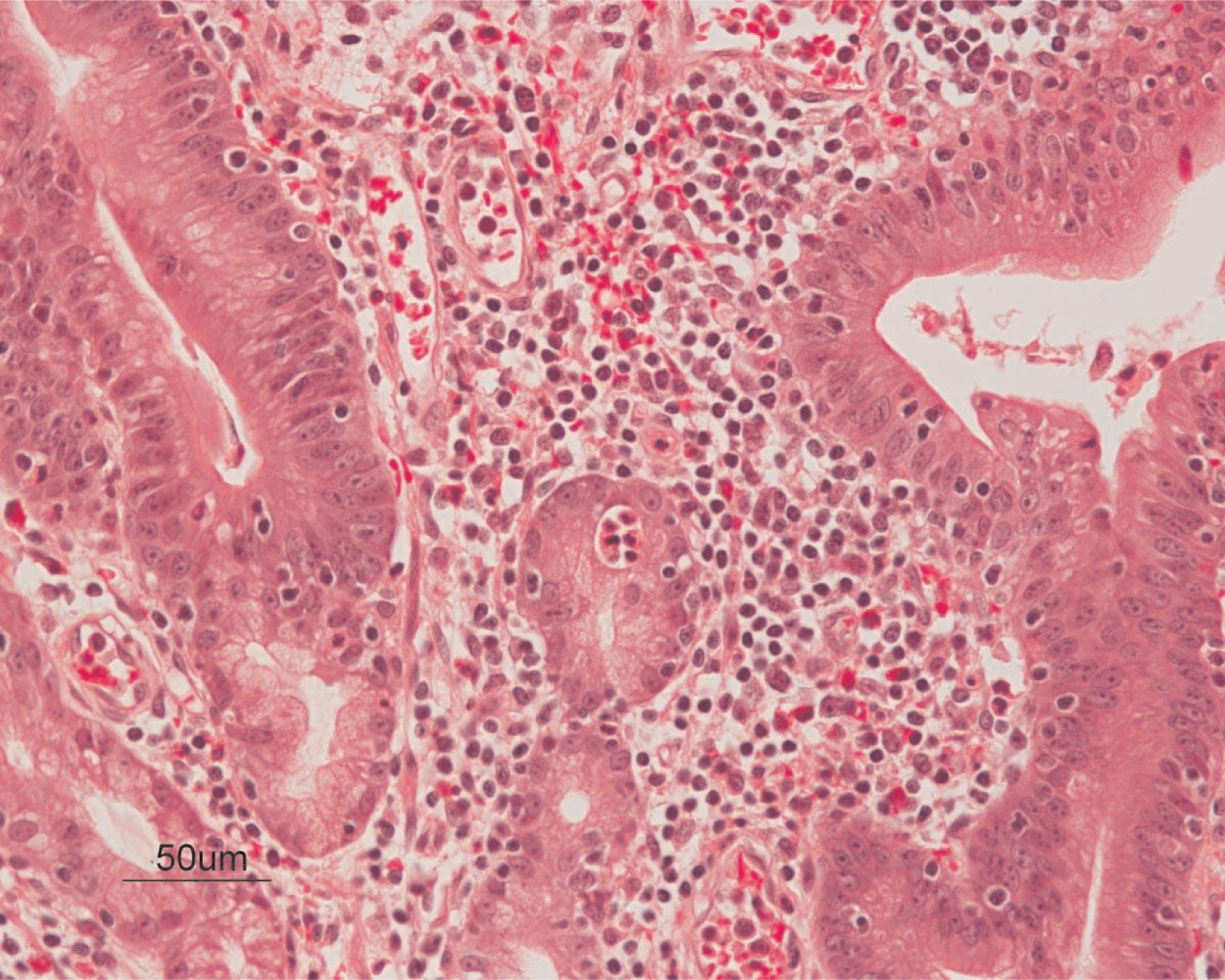
Figure 5:
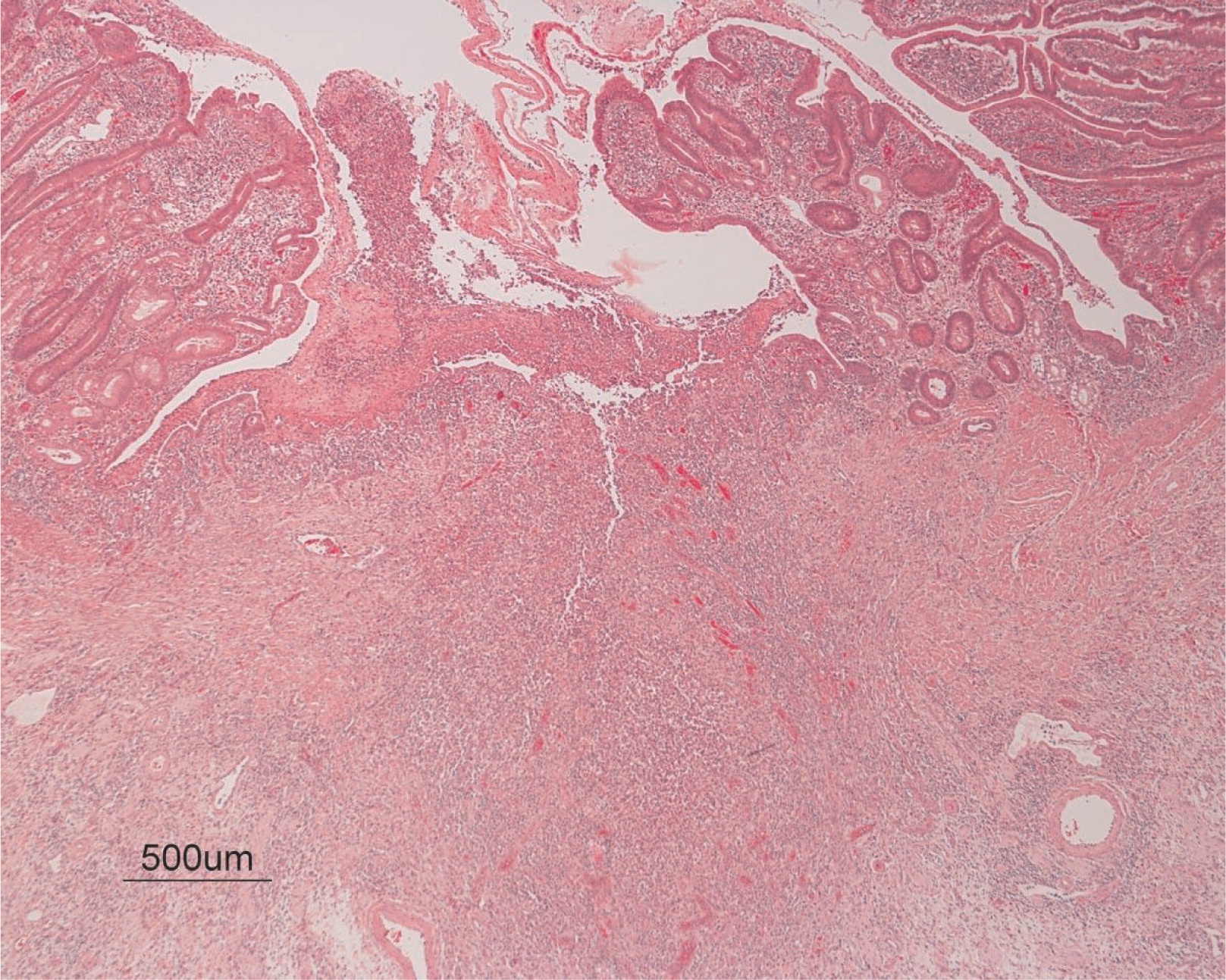
Figure 6:
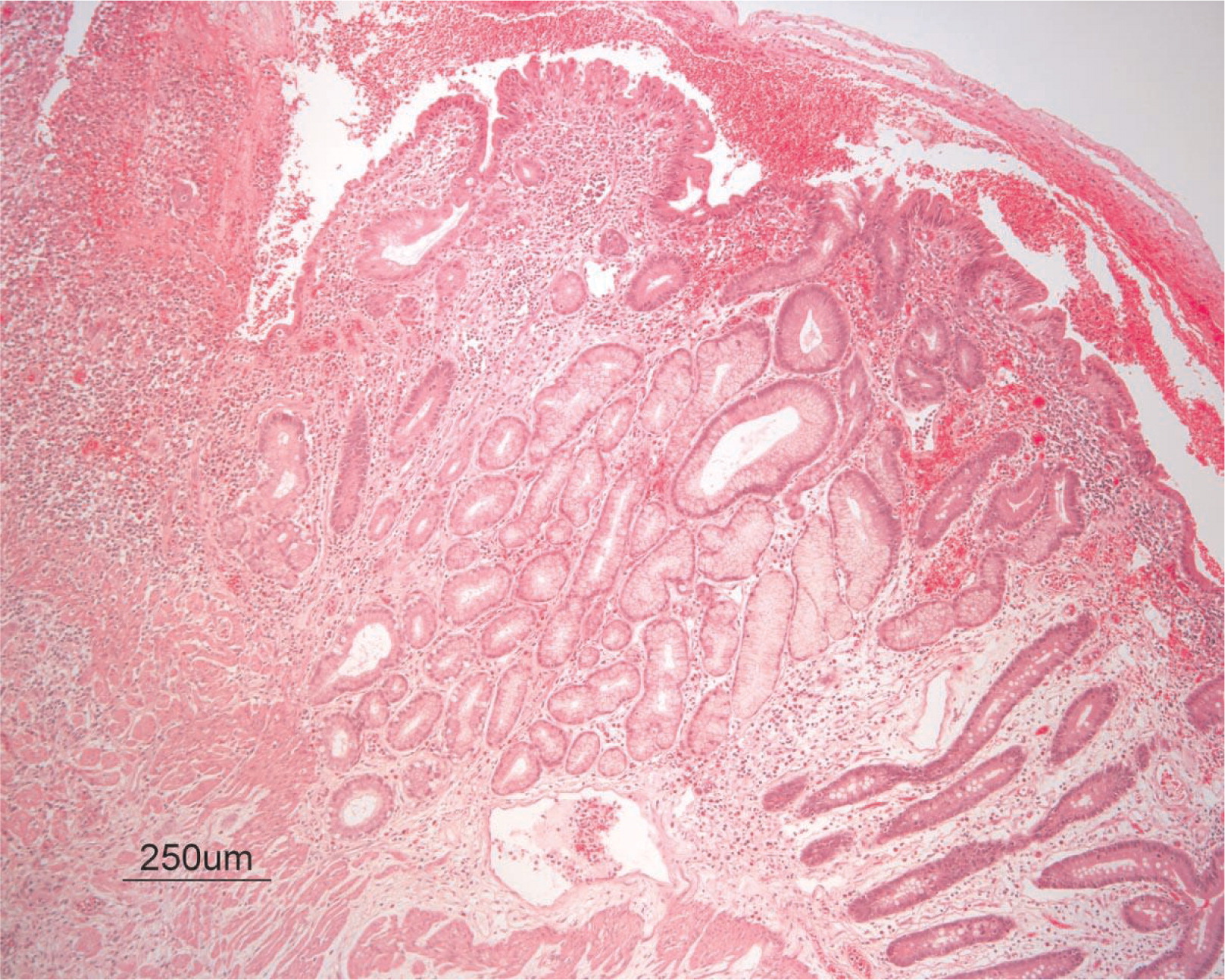
Figure 7:
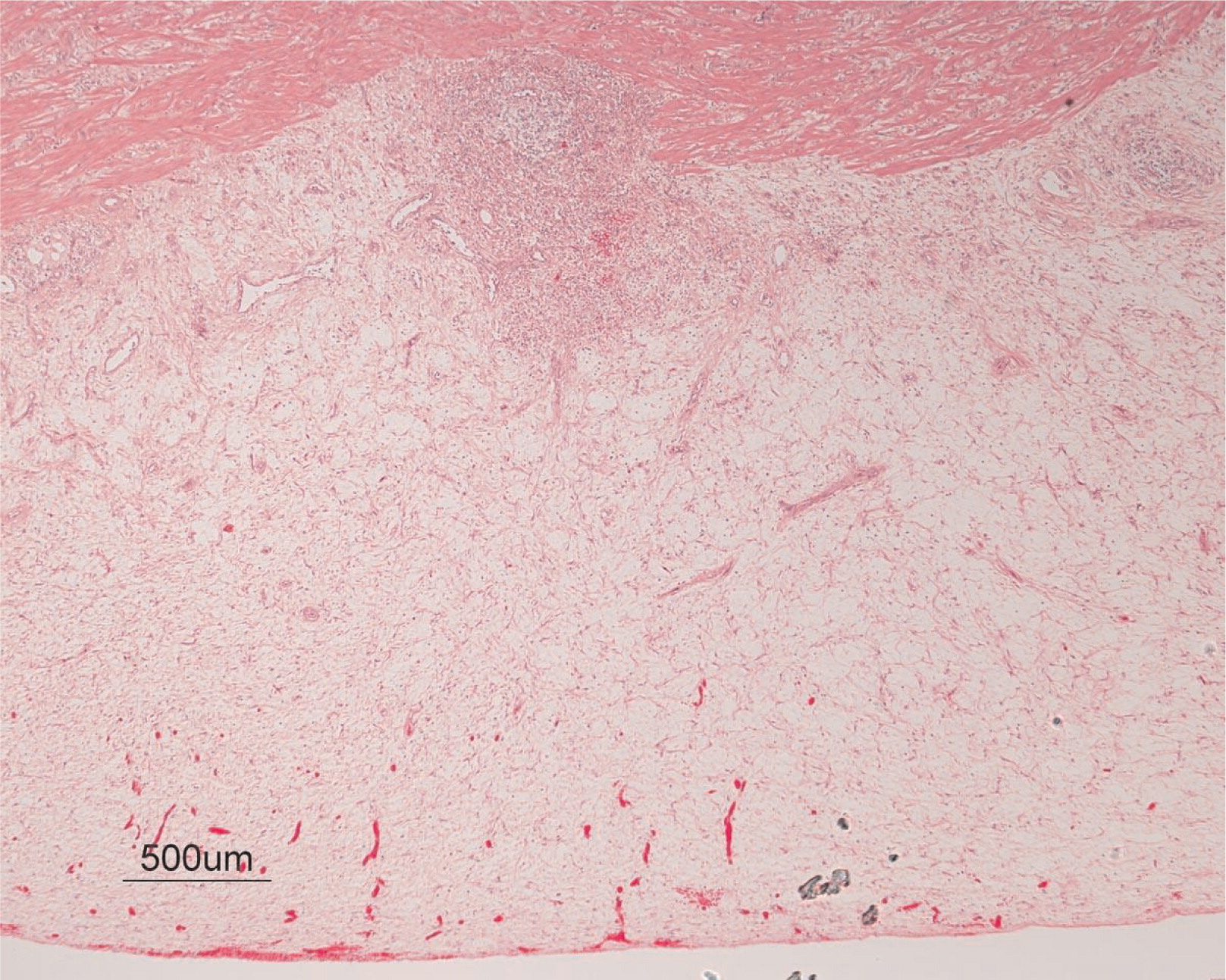
Figure 8:
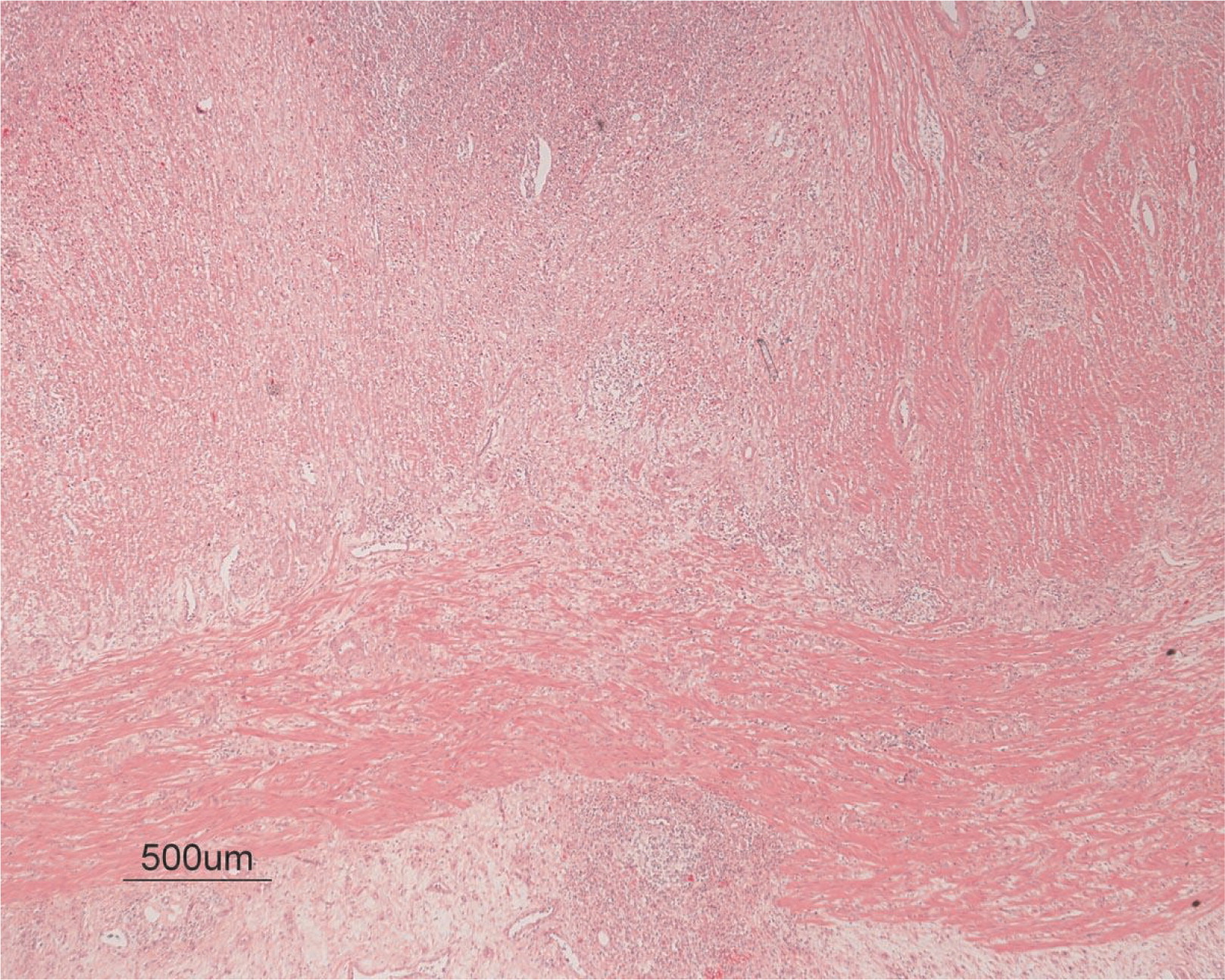
Figure 9:
Following the resection, a complete resolution of the symptoms including protein-losing enteropathy ensued. Ten years later he remained completely symptom-free from GI complaints.
Immune evaluation
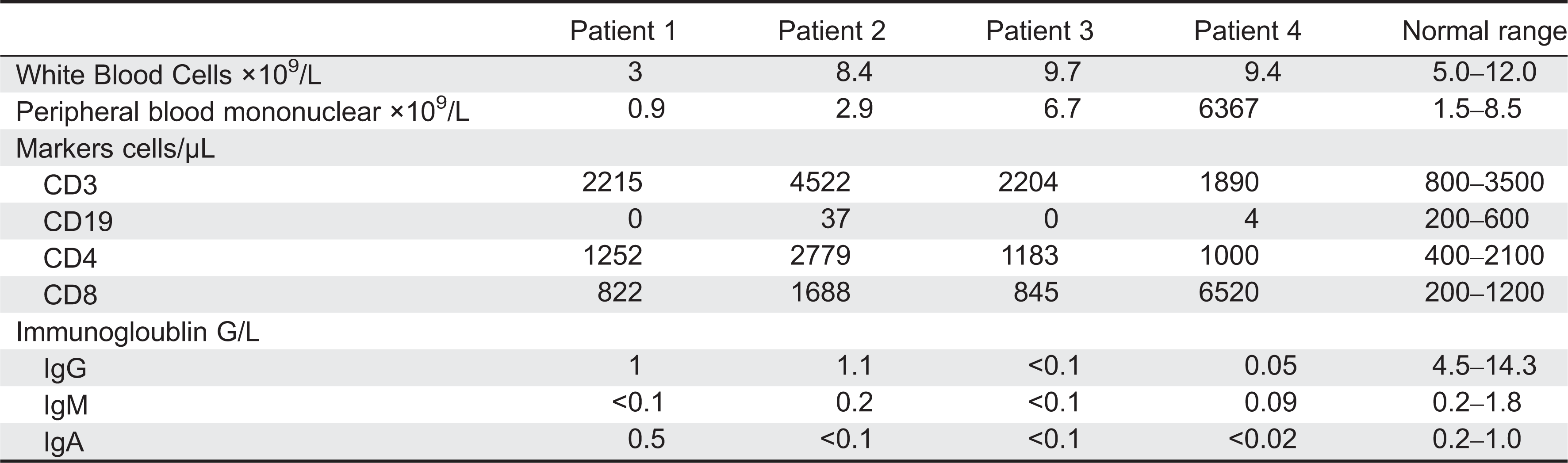
All patients but patient 1, who had neutropenia, had normal numbers of circulating white blood cells. Lymphocyte numbers were normal with normal distribution of CD3+ CD4+ as well as CD3+ CD8+ cells. In contrast, CD19+ B-cell numbers were universally low to absent.
Immunoglobulin levels were also almost universally depressed. IgG was extremely low in all patients. IgM was borderline in patient 2, but it was depressed in all other patients. IgA was normal in patient 1, but it was depressed in all other patients (Table 1).
Table 1:
Genetic analysis
Despite the unusual clinical presentation, the low number of circulating B cells as well as the low levels of immunoglobulin prompted the sequence analysis of the BTK gene in these patients. One patient had nonsense mutation, whereas 3 other patients had missense mutations (Table 2).
Table 2:

Patient 1 had a C to T transition at position 1921, which predicts an arginine to cysteine substitution at position 641 in the kinase domain.
Patient 2 had a T to C exchange at position 98, which predicts a phenylalanine to leucine substitution.
Patient 3 had a T to G transition at position 279, creating a stop codon in the SH3 domain of BTK.
Patient 4 had a T to C transition at position 925 resulting in a serine to proline exchange at position 309 in the SH2 domain.
Discussion
Typically, XLA presents in infancy or childhood with repeated microbial infections and a complete lack of tonsils and lymph nodes. However, several cases with atypical presentation in adult life have been reported. In our cohort of patients with XLA we have identified 4 patients with unusual and unique presentations or manifestations. Invasive infections were uncommon, responded well to antibiotic treatment, or resolved after myringotomy tube insertion. Such features are common in the pediatric population and therefore did not trigger an assessment for primary immunodeficiency. In addition, small cervical lymph nodes were present in some cases, which may have misled the primary care physician and ear, nose, and throat specialist.
The main presentation from patient 1 was unusual granulomatous skin eruptions. Granulomatous Dermatitis (GD) is defined by an infiltration of mononuclear and multinucleate giant cells in the skin, with or without lymphocyte infiltration (Mohan et al. 2006). There are several histopathological types of granulomatous reactions in the skin and they include tuberculoid, sarcoid, necrobiotic, suppurative, foreign bodies, and other rare causes such as primary or secondary immunodeficiencies. In infants and young children GD may be associated with infectious caused by mycobacteria, bacteria, fungi, or viral agents (Goodless et al. 1991; Gibney et al. 1996; Eisendle and Zelger 2009), and it may be linked to various medications such as calcium channel blockers, ACE inhibitors, and others (Magro et al. 1998). In older children and adolescents, it can be associated with autoimmune diseases such as sarcoidosis, rheumatoid arthritis, systemic lupus erythematous (Rose et al. 2014), or inflammatory bowel disease. GD has also been described in immunodeficiency states such as chronic granulomatous disease and common variable immunodeficiency (CVID) (Torrelo et al. 1995). None of these associations could be identified in our patients. It therefore remains unclear why this patient was afflicted with GD. It is possible that this aberrant inflammatory process might be related to effects stemming from BTK dysfunction in a variety of cell types such as monocytes and dendritic cells (Koprulu and Ellmeier 2009). XLA patients have a high percentage of CD14+CD16+ and CD14 CD35+ cells, but phagocytosis and chemotaxis by these cells seems reduced (Amoras et al. 2007). Moreover, monocytes from BTK-deficient patients showed an increased secretion of interleukin (IL)-6, IL-10, and tumor necrosis factor (TNF)-α in response to TLR 4, 7, and 8 stimulation (Marron et al. 2012), whereas cytokine secretion upon stimulation of dendritic cells following stimulation of TLR9 was significantly reduced (Lougaris et al. 2014). All these abnormalities that resulted in distorted inflammatory responses may have contributed to the formation of these granulomas in the patients with XLA.
Acute demyelinating encephalomyelitis (ADEM) is considered an infection related autoimmune demyelinating disease of the central nervous system. Pathogenesis of the disease is probably autoimmune and there are several proposed mechanisms. One leading theory is based on the assumption of molecular mimicry, involved structural, or partial amino-acid sequence homologies, which is shared between a pathogen and a host central nervous system (CNS) protein. In such a scenario, initial immune response to the pathogen leads to T-cell activation, which in turn cross-activates antigen-specific B cells creating auto-reactive T and B cells. These cells are capable of entering the CNS) and may recognize a homologous myelin protein (even long after clearance of the pathogen) eliciting an inflammatory reaction.
Another explanation might be a post-infectious etiology, whereby a direct CNS infection causes tissue damage and a disruption of the blood–brain barrier. This may lead to leakage of CNS-privileged auto antigens that could be processed in systemic lymphatic organs, eliciting a self-reactive encephalopathic T-cell response. In patient 2 a viral illness may have led to development of this severe sequel. Although viral meningoencephalitis due to ECHO and other viruses is a common finding in XLA patients, the infection-related or autoimmune encephalitis such as ADEM is uncommon. This may have been due to an aberrant immune reaction owing to BTK deficiency. Although T cells are responsible for most antiviral responses, B cells and antibodies may be critical in the response to some viruses such as enteroviruses, hence the increased susceptibility of XLA to echovirus (Halliday et al. 2003). However, viruses were not detected in the CSF obtained from this patient. Nevertheless this possibility cannot be completely excluded.
Another unusual manifestation for XLA was idiopathic thrombocytopenic purpura (ITP), identified in patient 3. This is a common putatively autoimmune phenomenon in other immune deficiency states such as CVID and autoimmune lymphoproliferative syndrome but not in XLA. Using the common assumption that ITP is caused by auto antibodies, it was a surprising occurrence in a patient who has difficulties producing any antibodies. This may suggest some residual ability in XLA patients to produce antibodies. In fact, a short time after starting replacement with IVIg the thrombocytopenia resolved, thus supporting the possibility of an underlying autoimmune mechanism.
Finally, patient 4 had a prolonged course of Crohn's-like inflammatory bowel disease or infantile colitis with a prominent feature of protein-losing enteropathy. The bowel lesion was limited to a jejunal segment that upon resection led to complete resolution of all symptoms. Although the findings from the biopsies were showing inflammation, they were not of the granulomatous type and were thus incompatible with classic Crohn's disease. Chronic noninfectious inflammatory bowel disease manifesting as small intestine irregularities and strictures have been previously described in a few patients with XLA (Abramowsky and Sorensen 1988; Washington et al. 1996). With the recent discovery of several monogenic disorders such as FOXP3, IL-10, and IL-10R leading to infantile colitis, it is important to remember that XLA may present in a similar manner during infancy.
In this group of patients we saw manifestations that are atypical to XLA, in particular autoimmune phenomenon that are not known to be common in XLA patients.
Previous studies (López-Granados et al. 2005; Broides et al. 2006) suggested that a milder presentation may be related to missense mutations. In our group of patients we observed mutations in all regions of the gene with both missense and nonsense mutations. Despite having a nonsense mutation, patient 3 had a relatively mild course including a rash and later ITP. The small number of patients in this group precludes accurate statistical analysis, but it supports the previous notion of no clear genotype–phenotype correlation in XLA patients.
In summary, these 4 patients with novel mutations had a variety of unique inflammatory and autoimmune clinical manifestations that should alert physicians and immunologists to the possibility of XLA in similar cases.
REFERENCES
Abramowsky C.R. and Sorensen R.U. Regional enteritis-like enteropathy in a patient with agammaglobulinemia: histologic and immunocytologic studies Hum. Pathol. 1988 19 4 483 -486
Amoras A.L., da Silva M.T., Zollner R.L., Kanegane H., Miyawaki T., and Vilela M.M. Expression of Fc gamma and complement receptors in monocytes of X-linked agammaglobulinaemia and common variable immunodeficiency patients Clin. Exp. Immunol. 2007 150 3 422 -428
Broides A., Yang W., and Conley M.E. Genotype/phenotype correlations in X-linked agammaglobulinemia Clin. Immunol. 2006 118 2–3 195 -200
Bruton O.C. Agammaglobulinemia Pediatrics. 1952 9 722 -728
Eisendle K. and Zelger B. The expanding spectrum of cutaneous borreliosis G. Ital. Dermatol. Venereol. 2009 144 2 157 -171
Gibney M.D., Nahass G.T., and Leonardi C.L. Cutaneous reactions following herpes zoster infections: report of three cases and a review of the literature Br. J. Dermatol. 1996 134 3 504 -509
Goodless D.R., Ramos-Caro F.A., and Flowers F.P. Reactional states in Hansen's disease: practical aspects of emergency management South Med. J. 1991 84 2 237 -241
Halliday E., Winkelstein J.A., and Webster A.D. Enteroviral infections in primary immunodeficiency: a survey of morbidity and mortality J. Infect. 2003 46 1 1 -8
Hashimoto S., Miyawaki T., Futatani T., Kanegane H., Usui K., Nukiwa T., Namiuchi S., Matsushita M., Yamadori T., Suemura M., Kishimoto T., and Tsukada S. Atypical X-linked agammaglobulinemia diagnosed in three adults Intern. Med. 1999 38 9 722 -725
Koprulu A.D. and Ellmeier W. The role of Tec family kinases in mononuclear phagocytes Crit. Rev. Immunol. 2009 29 4 317 -333
Lin M.T., Chien Y.H., Shyur S.D., Huang L.H., Chiang Y.C., Wen D.C., and Liang P.H. Yang HCDe novo mutation in the BTK gene of atypical X-linked agammaglobulinemia in a patient with recurrent pyoderma Ann. Allergy Asthma. Immunol. 2006 96 5 744 -748
López-Granados E., Pérez de Diego R., Ferreira Cerdán A., Fontán Casariego G., and García Rodríguez M.C. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia J. Allergy Clin. Immunol. 2005 116 3 690 -697
Lougaris V., Baronio M., Vitali M., Tampella G., Cattalini M., Tassone L., Soresina A., Badolato R., and Plebani A. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation J. Allergy Clin. Immunol. 2014 133 6 1644 -1650
Magro C.M., Crowson A.N., and Schapiro B.L. The interstitial granulomatous drug reaction: a distinctive clinical and pathological entity J. Cutan. Pathol. 1998 25 2 72 -78
Marron T.U., Martinez-Gallo M., Yu J.E., and Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines J. Allergy Clin. Immunol. 2012 129 1 184 -190
Mohan H., Bal A., and Dhami G.P. Non-infectious granulomatous dermatitis: a clinicopathological study J. Cutan. Pathol. 2006 33 12 767 -771
Rose C.D., Neven B., and Wouters C. Granulomatous inflammation: the overlap of immune deficiency and inflammation Best Pract. Res. Clin. Rheumatol. 2014 28 2 191 -212
Saulsbury F.T., Bernstein M.T., and Winkelstein J.A. Pneumocystis carinii pneumonia as the presenting infection in congenital hypogammaglobulinemia J. Pediatr. 1979 95 4 559 -561
Stewart D.M., Tian L., and Nelson D.L. A case of X-linked agammaglobulinemia diagnosed in adulthood Clin. Immunol. 2001 99 1 94 -99
Torrelo A., Mediero I.G., and Zambrano A. Caseating cutaneous granulomas in a child with common variable immunodeficiency Pediatr. Dermatol. 1995 12 2 170 -173
Washington K., Stenzel T.T., Buckley R.H., and Gottfried M.R. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia Am. J. Surg. Pathol. 1996 20 10 1240 -1252
Winkelstein J.A., Marino M.C., Lederman H.M., Jones S.M., Sullivan K., Burks A.W., Conley M.E., Cunningham-Rundles C., and Ochs H.D. X-linked agammaglobulinemia: report on a United States registry of 201 patients Med. (Baltimore). 2006 85 4 193 -202
Information & Authors
Information
Published In

LymphoSign Journal
Volume 2 • Number 2 • June 2015
Pages: 75 - 83
History
Received: 6 November 2014
Accepted: 11 November 2014
Accepted manuscript online: 11 November 2014
Version of record online: 11 November 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Amit Nahum, David Manson, and Bo Ngan. 2015. Atypical presentation and manifestations in X-linked agammaglobulinemia patients with novel BTK mutations. LymphoSign Journal.
2(2): 75-83. https://doi.org/10.14785/lpsn-2014-0015
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
