Chronic granulomatous disease with an initial presentation of arthritis and oral ulcers
Abstract
Chronic granulomatous disease (CGD) is an inherited defect of leukocyte phagocytic function leading to recurrent infections. Autoimmune manifestations are reported in up to 6% of patients with CGD. We report a case of CGD presenting with arthritis as the first manifestation of disease. A 12-year-old Pakistani male of consanguineous parents presented with migratory arthritis and painless oral ulcerations of 6 months duration that were minimally responsive to nonsteroidal anti-inflammatory treatment. Initial assessment demonstrated elevated inflammatory markers (ESR 62), weakly positive ANA (titer 1:40), negative anti-DsDNA, and negative RF. He presented to the emergency department with fevers and arthritis. Repeat work-up suggested early Macrophage Activation Syndrome: normocytic anemia (Hgb 95 g/L), thrombocytopenia (Plt 141 × 109/L), elevated LDH 1603, ferritin 1230 mcg/L, ESR 127, CRP 9.3, hypertiglycerdemia (3.2 mmoL/L) and mild transaminitis (ALT 63, AST 87), normal bone marrow (no hemophagocytosis), but mildly elevated Soluble CD 136 (1086 ng/mL) and Soluble IL-2 receptor (CD25) (1698 U/mL). He was treated with oral prednisone with symptom resolution. The arthritis relapsed after 1 month and the patient developed fever, productive cough, and pleuritic chest pain. Chest imaging revealed multiple nodular opacities and enlarged mediastinal lymph nodes. Aspergillus fumigatus complex was isolated from induced sputum prompting screening for primary immunodeficiency. Neutrophil oxidative burst function, as assessed by a dihydrorhodamine flow cytometry based assay, was low at 1.26 and 1.48 (normal range 32–300). Genetic analysis showed a previously described mutation in the NCF1 gene confirming the diagnosis of autosomal-recessive CGD. CGD can present with an exclusively rheumatologic presentation including arthritis and oral ulceration.
Statement of Novelty: This case demonstrates that CGD can present with rheumatological symptoms prior to any infectious features.
Introduction
Chronic Granulomatous Disease (CGD) is a rare, inherited defect of leukocyte phagocytic function with an inability to produce superoxide radicals. It is the result of a mutation in at least 1 of 5 genes that encode nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex enzyme. Two of the protein subunits are membrane-bound glycoprotein gp91phox (phagocyte oxidase) and p22phox; the remaining subunits are cytosolic p47phox, p67phox, and p40phox (Segal et al. 2000).
The disease is characterized by recurrent fungal and bacterial infections, with a predilection to catalase-producing organisms. Other recognized disease manifestations include granulomata causing resultant gastrointestinal or urinary obstruction, inflammatory bowel disease, and rarely, autoimmune disease (Kang et al. 2011).
Autoimmune manifestations reported in CGD include Lupus Syndromes (Discoid Lupus Erythematous and Systemic Lupus erythematous), idiopathic/immune thrombocytopenia, myasthenia gravis, sarcoid-osis, IgA nephropathy, steroid responsive pericardial effusion, anti-phospholipids syndrome and autoimmune hepatitis (Schmitt et al. 1995; Macedo et al. 1999; Winkelstein et al. 2000; De Ravin et al. 2006, 2008; van den Berg 2009; Koker et al. 2013).
The prevalence of CGD differs by country with estimated prevalence up to 1:111 000 to 1:1 330 000 in Israeli Arabs and the United Kingdom to as low as 1:1 000 000 in a recent Italian cohort (Jones et al. 2008; Martire et al. 2008; Wolach et al. 2008). In North America, the reported disease prevalence is 1:200 000–250 000 with 70% of patients having the mutation in the X-linked CYBB gene encoding gp91phox. Other gene mutations are autosomal recessive (AR); the most common AR gene affected is neutrophil cytosolic factor 1 (NCF1), encoding p47phox (Winkelstein et al. 2000). In the Middle East, most patients have autosomal recessive mutations as opposed to the X-linked CYBB gene (Fattahi et al. 2011; Al-Muhsen and Alsum 2012; Koker et al. 2013).
Functional and clinical presentation
A 12-year-old Pakistani male of consanguineous parents presented with migratory arthritis and painless oral ulcerations of 6 months duration. The arthritis involved metacarpopharyngeal (MCP), metatarsopharyngeal (MTP), and proximal interphalangeal (PIP) joints at various time points. However, within a week, the patient had diffuse arthritis in most MCP, MTP, and PIP joints of the hands and feet. He had accompanying arthralgias in his neck, shoulder, elbows, wrists, and knees. Painless oral ulcerations were swabbed with negative results for viruses. The arthritis resolved within a week with a nonsteroidal anti-inflammatory (ibuprofen) and without the use of antimicrobial drugs.
Four months later, his arthritis relapsed and he developed daily fevers, anorexia, fatigue, and weight loss. Blood work showed anemia (Hgb 110 g/L), platelets (172 × 109/L), white blood cells (5.2 × 109/L), and differential (neutrophils 2.5 × 109/L, lymphocytes 2.2 × 109/L). Inflammatory markers were elevated (ESR 62, CRP 29). Rheumatological work-up included a weakly positive antinuclear antibody (ANA) (titer 1:40), negative staining for anti-DsDNA, anti-RNP, anti-Smith, anti-cardiolipin, anti-SS-A, anti-SS-B, and rheumatoid factor. He had transient response to naproxen. His fever persisted and he was found to have angular stomatitis, cervical lymphoadenopathy, hepatomegaly, and arthritis in the left PIP joints of the hand and in the right ankle. Repeated blood work was significant for a normocytic anemia (Hgb 95 g/L), thrombocytopenia (Plt 141 × 109/L), elevated LDH 1603 U/L, ferritin 1230 mcg/L, ESR 127, CRP 9.3, hypertiglycerdemia (3.2 mmoL/L), and mild transaminitis (ALT 63, AST 87). Bone marrow aspirate was not suggestive of hemophagocytosis. Mildly elevated soluble CD 136 (1086 ng/mL) and soluble IL-2 receptor (CD25) (1698 U/mL) were noted. The patient was treated for presumptive Macrophage Activation Syndrome given his arthritis history and blood work. He received 60 mg of oral prednisone with complete symptom resolution, and he was discharged on a tapering schedule. Again, no antimicrobial agents were used.
After 1 month, the arthritis and fever relapsed, and the patient developed a new productive cough and pleuritic chest pain. Chest imaging illustrated multiple nodular opacities and enlarged mediastinal lymph nodes (Figure 1). Induced sputum demonstrated the presence of yeast and pseudohyphae. A follow-up bronchoalveolar lavage confirmed Aspergillus fumigatus complex. The identification of this unusual infection prompted a request for an Immunology consultation.
Figure 1:
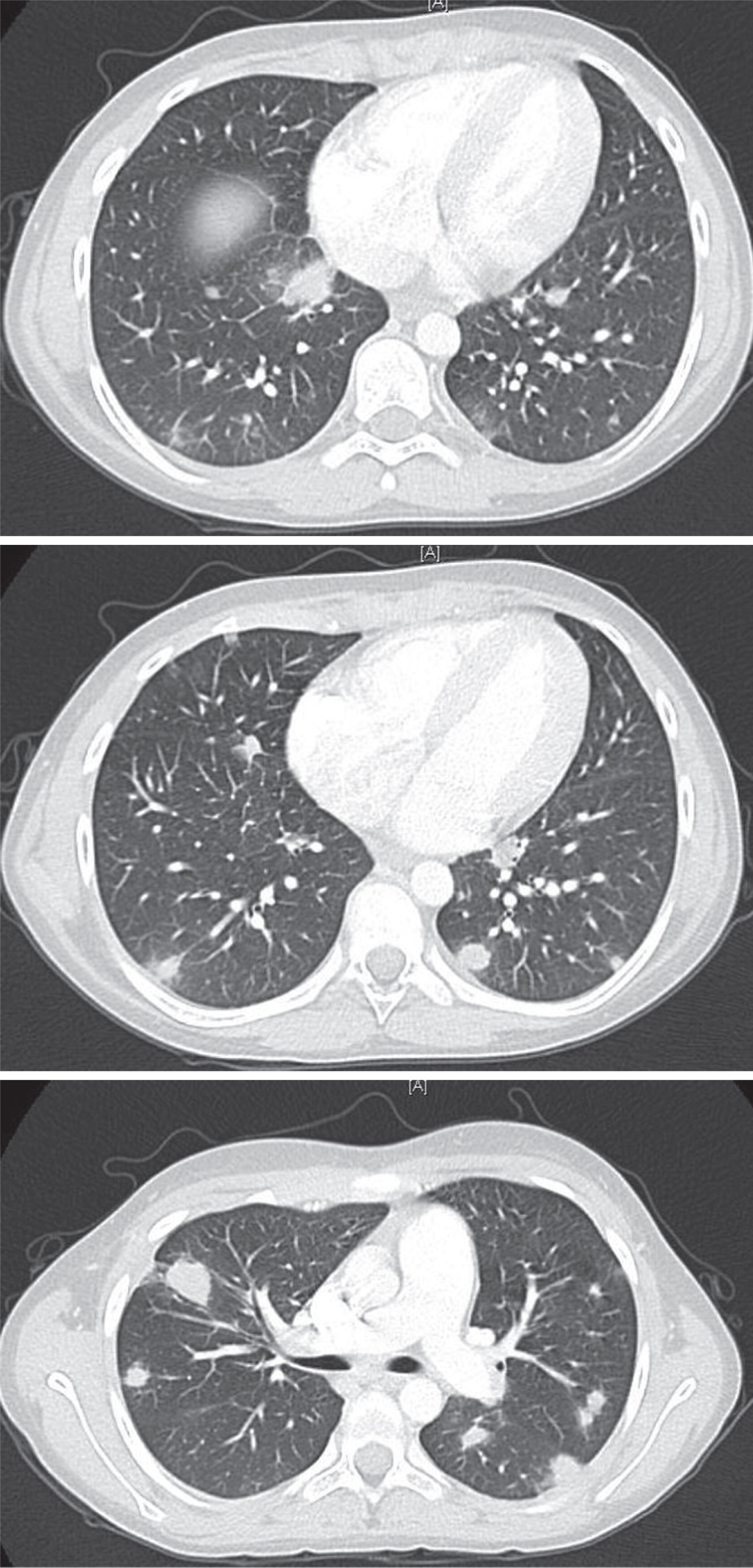
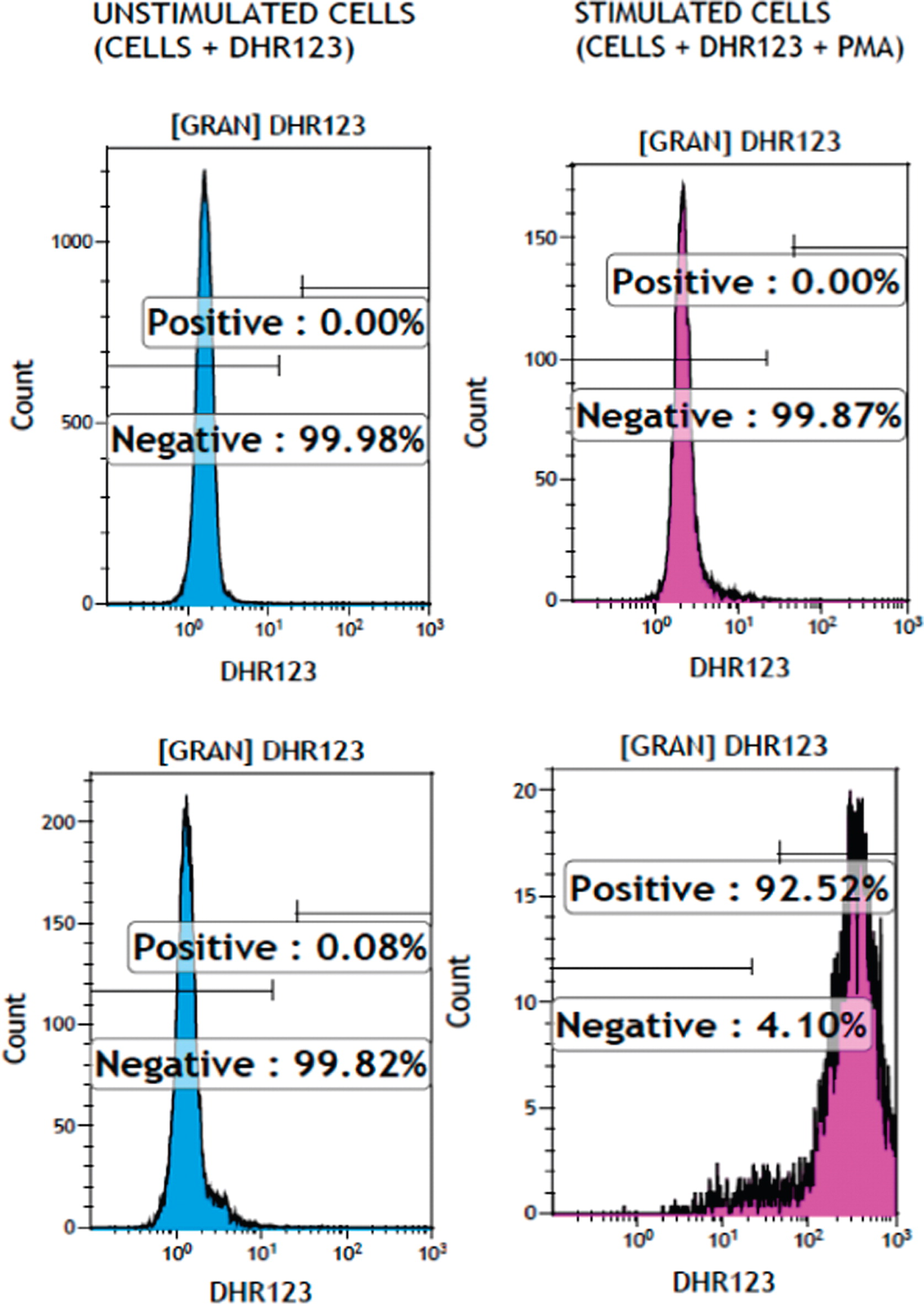
Immunological assessment found that his past medical history was positive for uncomplicated varicella disease at 5 years of age. There was no previous history of pneumonia, lymphadenitis, deep-seated abscess, or osteomyelitis and no prior history of recurrent abdominal pain, vomiting, diarrhea, or blood in the stool. His immunization record was up to date including Bacillus Calmette–Guerin vaccine with no adverse reactions or complications. He had unremarkable lymphocytes subsets (CD3+: 2511 (normal range 800–3500), CD4+: 1250 (normal range 400–2100), CD8+: 1123 (normal range 200–1200), CD19+: 1014 (normal range 200–600), and NK cells: 36 (normal range 70–1200), all in cells/µL), high IgG 19.2 g/L (normal range 6.60–15.30 g/L), high IgE 294 IU/mL (normal range < 200 IU/mL), and normal IgM 1.5 g/L (normal range 0.40–1.5 g/L) and IgA 2.9 g/L (normal range 0.50–2.2 g/L). Specific antibody responses were positive for measles, mumps, rubella and varicella, and protective tetanus titers (0.55 IU/mL). Owing to the finding of Aspergillus fumigatus, neutrophil oxidative burst function as assessed by dihydrorhodamine flow cytometry based assay (Figure 2) was abnormally low at 1.26 and 1.48 (normal range 32–300). Genetic testing confirmed a homozygous mutation in the NCF1 gene (encoding for p47phox) at c.75_76delGT. The patient received treatment for lung aspergillosis with voriconazole and was placed on cotrimoxazole bacterial prophylaxis. On follow-up, he has continued these treatments and has shown signs of improvement in the size of the lung lesions. He developed intermittent abdominal pain without diarrhea. Enterography MRI and upper and lower endoscopies including biopsies were all normal.
Figure 2:
Discussion
The NADPH oxidase complex enzyme is critical in microbial killing, but it also serves a protective role in inflammation and autoimmunity (Brown et al. 2003; Hultqvist et al. 2009). CGD patients are at an increased risk of colitis and discoid lupus, in addition to arthritis, although the latter is rarely reported (Winkelstein et al. 2000; De Ravin et al. 2008; van den Berg et al. 2009). There are case reports as well as entries in large registries that have previously reported arthritis developing after the diagnosis of CGD (Lee and Yap 1994; De Ravin et al. 2008; van den Berg et al. 2009; Koker et al. 2013; Magnani et al. 2014). There are 7 patients reported with arthritis, and only 3 with systemic lupus erythematosus (SLE), found in various large registries of CGD patients. Gene defects are however not reported in these patients and the phenotype of the arthritis is not described (Table 1).
Table 1:
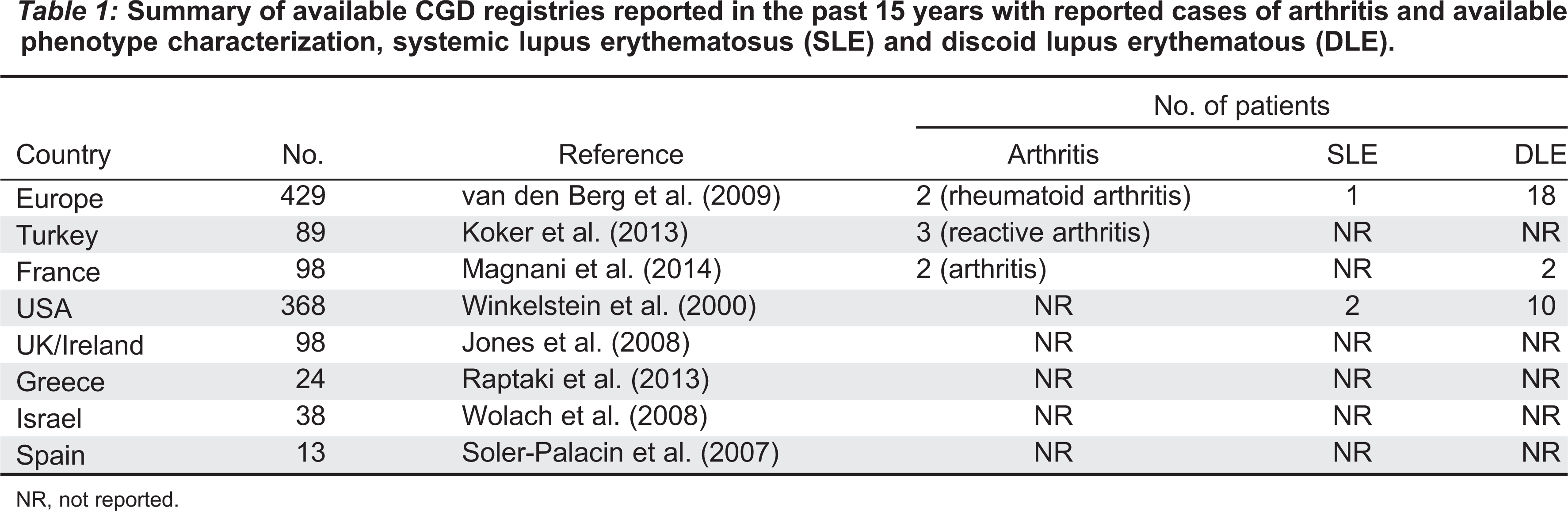
NR, not reported.
Our patient’s mutation (c.75_76delGT) accounts for greater than 95% of mutations in the NCF1 gene (Noack et al. 2001). The NCF1 gene encodes for the p47phox subunit of the NADPH oxidase complex. Deficiency in the p47phox subunit has been reported in 2 case reports of CGD patients with arthritis. Both of these patients developed arthritis after the diagnosis of CGD. The first case was diagnosed with p47phox deficiency at 2 years of age after presenting with Aspergillus pneumonia (Lee and Yap 1994). She developed a diffuse polyarthritis at 4 years of age. Rheumatological evaluation was significant for an elevated rheumatoid factor 395 IU/mL (normal 0–30), weakly positive ANA (1:40, homogenous patter), and positive anti-dsDNA (11 mg/mL). There was also radiographic evidence of progressive bony erosions.
The second case was diagnosed with p47phox deficiency at birth (De Ravin et al. 2008). Her early course was remarkable for recurrent sinusitis, otitis media, and pulmonary infections. She developed a polyarthritis at the 15 years of age with multiple rheumatoid nodules. Rheumatological work-up demonstrated rheumatoid factor in the synovial fluid, and biopsy showed perivascular synovitis without lymphoid aggregates. Imaging illustrated mild subchondreal lucency. The patient required Etanercept for full symptom resolution.
Animal models support the importance of the oxidative burst pathway and the NCF1 gene in particular in the pathogenesis of arthritis. Rodents with a homogenous mutation in the NCF1 gene develop severe autoimmune arthritis (Hultqvist et al. 2004, 2009).
There are various proposed mechanisms for the development of hyperinflammation and autoimmunity in patients with CGD. Animal and human cells exhibit reduced NADPH oxidase activity required for the induction of T regulatory cells (Magnani et al. 2014). Other proposed mechanisms have recently been reviewed (Rieber et al. 2012). These mechanisms include a reduction in neutrophil apoptosis, an imbalance of innate immune receptors (toll-like and complement receptors), alterations in T-cell activation and proliferation due to altered reduction of thiol groups on the T-cell membrane, induction of Th17 cells, impaired Nrf2 activity (a key redox-sensitive anti-inflammatory regulator), and increased inflammasome activation. However, the clinical importance of many of these mechanisms requires further investigation.
Our patient is rare because not only did he have arthritis, which is rare in CGD, but he also had this presentation prior to any other manifestations of the disease. Clinically, the course of arthritis in our patient was likely of autoimmune origin and very unlikely to be of infectious etiology given the intermittent nature of his arthritis, the prompt response to anti-inflammatory drugs, and the complete resolution prior to introducing antimicrobial treatment.
His arthritis was nondestructive; it was diffuse with symmetrical involvement of the small joints of the hands and feet (PIPs, MCPs, and MTPs). Large joints were less affected. The patient was evaluated for SLE. There were neither symptoms nor signs suggestive of photosensitivity, malar or discoid rash, serositis, renal or neurological involvement, as well as negative autoantibodies with the exception of a weakly positive ANA (1:40). He did have anemia with intermittent reticulocytosis and oral ulcers. Therefore, he had only 4 possible criteria of SLE including a borderline ANA. Given that some of his features, such as the oral ulcers (van den Berg et al. 2009), can be explained by his CGD, he does not currently carry a diagnosis of SLE.
Steroid use is a known risk factor for invasive Aspergillus infections, which is positively correlated to the dose and duration (Baddley 2011). In this case, the duration of steroids was short and was not likely to be sufficient to render him prone to this infectious complication. In patients with rheumatological disease and unusual infectious organisms the possibility of a primary immunodeficiency disorder (PID) is plausible (Todoric et al. 2013).
Aspergillus is a common infectious complication of CGD. A detailed discussion of the management of CGD is beyond the scope of this article and has been reviewed recently by Kang et al. (2011). In brief, the management includes prophylactic antifungal medications against Aspergillus, antibacterial prophylaxis against Staphylococcus Aureus, and consideration of hematopoietic stem cell transplantation.
Conclusions
This case illustrates that rheumatological features can be the presenting feature of a PID and not just an associated feature. It also emphasizes the need to consider a PID in patients with rheumatologic diseases who develop unusual or opportunistic infections. Increased physician awareness may lead to more recognition of the autoimmune manifestations of CGD.
Acknowledgements
Dr. Mohammad Alsalamah is also sponsored by King Saud University for Health Science, Riyadh, Saudi Arabia.
REFERENCES
Al-Muhsen S. and Alsum Z. Primary immunodeficiency diseases in the Middle East Ann. N. Y. Acad. Sci. 2012 1250 56 -61
Baddley J.W. Clinical risk factors for invasive aspergillosis Med. Mycol. 2011 49 Suppl 1 S7 -S12
Brown J.R., Goldblatt D., Buddle J., Morton L., and Thrasher A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) J. Leukoc. Biol. 2003 73 591 -599
De Ravin S.S., Naumann N., Cowen E.W., Friend J., Hilligoss D., Marquesen M., Balow J.E., Barron K.S., Turner M.L., Gallin J.I., and Malech H.L. Chronic granulomatous disease as a risk factor for autoimmune disease J. Allergy Clin. Immunol. 2008 122 1097 -1103
De Ravin S.S., Naumann N., Robinson M.R., Barron K.S., Kleiner D.E., Ulrick J., Friend J., Anderson V.L., Darnell D., Kang E.M., and Malech H.L. Sarcoidosis in chronic granulomatous disease Pediatrics. 2006 117 e590 -e595
Fattahi F., Badalzadeh M., Sedighipour L., Movahedi M., Fazlollahi M.R., Mansouri S.D., Khotaei G.T., Bemanian M.H., Behmanesh F., Hamidieh A.A., Bazargan N., Mamishi S., Zandieh F., Chavoshzadeh Z., Mohammadzadeh I., Mahdaviani S.A., Tabatabaei S.A., Kalantari N., Tajik S., Maddah M., Pourpak Z., and Moin M. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease J. Clin. Immunol. 2011 31 792 -801
Hultqvist M., Olofsson P., Holmberg J., Backstrom B.T., Tordsson J., and Holmdahl R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene Proc. Natl. Acad. Sci. U. S. A. 2004 101 12646 -12651
Hultqvist M., Olsson L.M., Gelderman K.A., and Holmdahl R. The protective role of ROS in autoimmune disease Trends. Immunol. 2009 30 201 -208
Jones L.B., McGrogan P., Flood T.J., Gennery A.R., Morton L., Thrasher A., Goldblatt D., Parker L., and Cant A.J. Special article: Chronic granulomatous disease in the United Kingdom and Ireland: A comprehensive national patient-based registry Clin. Exp. Immunol. 2008 152 211 -218
Kang E.M., Marciano B.E., DeRavin S., Zarember K.A., Holland S.M., and Malech H.L. Chronic granulomatous disease: Overview and hematopoietic stem cell transplantation J. Allergy Clin. Immunol. 2011 127 1319 -1326; quiz 1327–1328
Koker M.Y., Camcioglu Y., van Leeuwen K., Kilic S.S., Barlan I., Yilmaz M., Metin A., de Boer M., Avcilar H., Patiroglu T., Yildiran A., Yegin O., Tezcan I., Sanal O., and Roos D. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients J. Allergy Clin. Immunol. 2013 132 1156 -1163. e5
Lee B.W. and Yap H.K. Polyarthritis resembling juvenile rheumatoid arthritis in a girl with chronic granulomatous disease Arthritis Rheum. 1994 37 773 -776
Macedo F., McHugh K., and Goldblatt D. Pericardial effusions in two boys with chronic granulomatous disease Pediatr. Radiol. 1999 29 820 -822
Magnani A., Brosselin P., Beaute J., de Vergnes N., Mouy R., Debre M., Suarez F., Hermine O., Lortholary O., Blanche S., Fischer A., and Mahlaoui N. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease J. Allergy Clin. Immunol. 2014 134 655 -662. e8
Martire B., Rondelli R., Soresina A., Pignata C., Broccoletti T., Finocchi A., Rossi P., Gattorno M., Rabusin M., Azzari C., Dellepiane R.M., Pietrogrande M.C., Trizzino A., Di Bartolomeo P., Martino S., Carpino L., Cossu F., Locatelli F., Maccario R., Pierani P., Putti M.C., Stabile A., Notarangelo L.D., Ugazio A.G., Plebani A., and De Mattia D. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: An Italian multicenter study Clin. Immunol. 2008 126 155 -164
Noack D., Rae J., Cross A.R., Ellis B.A., Newburger P.E., Curnutte J.T., and Heyworth P.G. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: Mutations not arising in the NCF-1 pseudogenes Blood. 2001 97 305 -311
Raptaki M., Varela I., Spanou K., Tzanoudaki M., Tantou S., Liatsis M., Constantinidou N., Bakoula C., Roos D., and Kanariou M. Chronic granulomatous disease: A 25-year patient registry based on a multistep diagnostic procedure, from the referral center for primary immunodeficiencies in Greece J. Clin. Immunol. 2013 33 1302 -1309
Rieber N., Hector A., Kuijpers T., Roos D., and Hart D. Current concepts of hyperinflammation in chronic granulomatous disease Clin. Dev. Immunol. 2012 2012 252460
Schmitt C.P., Scharer K., Waldherr R., Seger R.A., and Debatin K.M. Glomerulonephritis associated with chronic granulomatous disease and systemic lupus erythematosus Nephrol. Dial. Transplant. 1995 10 891 -895
Segal B.H., Leto T.L., Gallin J.I., Malech H.L., and Holland S.M. Genetic, biochemical, and clinical features of chronic granulomatous disease Medicine (Baltimore) 2000 79 170 -200
Soler-Palacin P., Margareto C., Llobet P., Asensio O., Hernandez M., Caragol I., and Espanol T. Chronic granulomatous disease in pediatric patients: 25 years of experience Allergol. Immunopathol. (Madr) 2007 35 83 -89
Todoric K., Koontz J.B., Mattox D., and Tarrant T.K. Autoimmunity in immunodeficiency Curr. Allergy Asthma Rep. 2013 13 361 -370
van den Berg J.M., van Koppen E., Ahlin A., Belohradsky B.H., Bernatowska E., Corbeel L., Espanol T., Fischer A., Kurenko-Deptuch M., Mouy R., Petropoulou T., Roesler J., Seger R., Stasia M.J, Valerius N.H., Weening R.S., Wolach B., Roos D., and Kuijpers T.W. Chronic granulomatous disease: The European experience PLoS One. 2009 4 e5234
Winkelstein J.A., Marino M.C., Johnston R.B. Jr, Boyle J., Curnutte J., Gallin J.I., Malech H.L., Holland S.M., Ochs H., Quie P., Buckley R.H., Foster C.B., Chanock S.J., and Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients Medicine (Baltimore) 2000 79 155 -169
Wolach B., Gavrieli R., de Boer M., Gottesman G., Ben-Ari J., Rottem M., Schlesinger Y., Grisaru-Soen G., Etzioni A., and Roos D. Chronic granulomatous disease in Israel: Clinical, functional and molecular studies of 38 patients Clin. Immunol. 2008 129 103 -114
Information & Authors
Information
Published In

LymphoSign Journal
Volume 3 • Number 1 • March 2016
Pages: 35 - 40
History
Received: 18 May 2015
Accepted: 19 July 2015
Accepted manuscript online: 30 September 2015
Version of record online: 30 September 2015
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Mohammad Alsalamah, Mariam Hanna, and Julia Upton. 2016. Chronic granulomatous disease with an initial presentation of arthritis and oral ulcers. LymphoSign Journal.
3(1): 35-40. https://doi.org/10.14785/lpsn-2015-0008
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
