Histamine, histamine receptors, and anti-histamines in the context of allergic responses
Abstract
Histamine is a bioactive amine which is considered a key player in the allergic response. Thus, histamine receptor blockers (antihistamines) play an important role in the treatment of a number atopic diseases such as allergic rhinitis, conjunctivitis, and acute and chronic forms of urticaria. Histamine is produced by immune cells but also by bacteria in the gut. Beyond its role in the acute allergic response, histamine exerts numerous effects by binding to its 4 pleiotropic G-protein coupled histamine receptors. Here, we describe the roles of these histamine receptors and antihistamines in the human system, clinical applications, side effects, and novel concepts for the usage of antihistamines with different specificity based on guidelines and recommendations.
Statement of novelty: This review provides an overview of histamine receptors and links it to clinical relevance of antagonizing their action in clinical routine.
Introduction
Histamine is a bioactive amine that acts as a signalling molecule and neurotransmitter. It exerts its diverse biological effects through the activation of 4 types of membrane bound receptors from the aminergic G-protein coupled receptor family: H1, H2, H3, and H4 (named according to their order of discovery). Due to their pleiotropic expression, these receptors can exert multiple clinical effects, but can also explain the side effects of therapeutics used to modify their action. Following the identification of histamine and H1 receptors, the therapeutic use of first generation H1 blockers (which lacked specificity) revealed a number of histamine related effects that were not inhibited. These cardiovascular, gastric (leading to peptic ulcer treatment discovery), neural, as well as various immune cell related mechanisms eventually led to the identification of additional histamine receptors as well and new molecules with agonist and antagonist effects. More recently, low-affinity intracellular non-H1, -H2, -H3, or -H4 receptors have been described in cell nuclei and microsomes, although the biological functions of these receptors are still somewhat unclear. This review focuses on the role of histamine receptors and anti-histamines in the context of allergic diseases.
Histamine in the context of atopic disease
Receptor structure
In general terms, the 4 histamine receptors are heptahelical G-protein coupled receptors (GPCR), encompassing a diverse group of membrane receptors composed of a single polypeptide that is folded into a globular shape, forming a 7-transmembrane structure. The extracellular loops are responsible for signalling molecule binding (N-terminal) (Bockaert and Pin 1999). Yamashita et al. (1991) cloned the structure of bovine H1 receptor, which is characterized by a large (212 amino acids) 3rd intracellular loop and a relatively short (17 amino acids) intracellular C-terminal tail. The third and fifth transmembrane domains of H1 and H2 receptors contribute to histamine binding (Birdsall 1991; Timmerman 1992). The full length human H1, H2, H3, and H4 receptors are composed of 487, 359, 445, and 390 amino acids, respectively (Simons 2004). H4 receptor has 40% homology with the H3 receptor, and was identified based on differences in tissue distribution and binding affinity (Nakamura et al. 2000). Liu et al. (2001) identified 35% homology between the H4 receptor and the H3 receptor (Table 1).
Table 1:
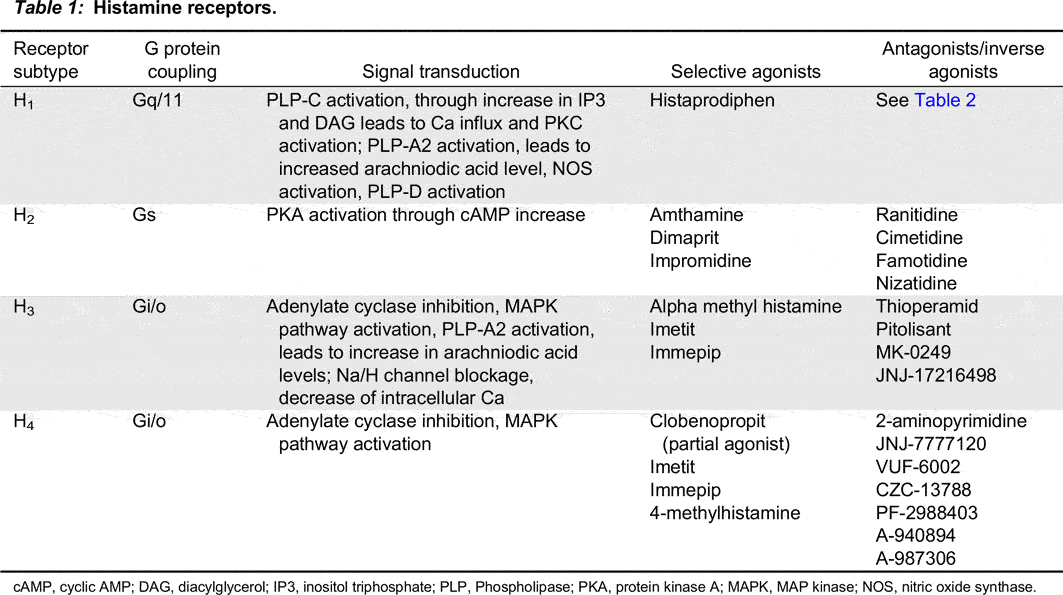
cAMP, cyclic AMP; DAG, diacylglycerol; IP3, inositol triphosphate; PLP, Phospholipase; PKA, protein kinase A; MAPK, MAP kinase; NOS, nitric oxide synthase.
Histamine receptor expression and function
The H1 receptor is widely distributed throughout the body, with well-documented expression in the central nervous system (CNS), smooth muscle, sensory nerves, heart, adrenal medulla, as well as immune, endothelial, and epithelial cells. It mediates most of the postsynaptic effects of histamine within the CNS. Through binding of the transmembrane domains 3 and 5 of H1 receptor, histamine stimulates smooth muscle contraction in the respiratory and gastrointestinal tract, stimulates sensory nerves leading to pruritus and sneezing, and increases vascular permeability (through prostacyclin, platelet activating factor, von Willebrand factor, and nitric oxide; NO) leading to oedema. H1 receptor intracellular signals are transmitted through Ca2+, cGMP, phospholipase D, phospholipase A2, and NF-κB activation. Simultaneous activation of H1 and H2 receptors via histamine can lead to clinical symptoms of anaphylaxis such as hypotension, tachycardia, flushing, and headache. With its diverse distribution and various effector activities, the H1 receptor is involved in allergic rhinitis, asthma, atopic dermatitis, conjunctivitis, and urticaria development.
The H2 receptor is widely expressed and can be found in gastric mucosal cells, heart, CNS, immune cells, and smooth muscles of the airway, vasculature, and uterus. It consists of a special short intracellular loop and long C-terminal tail. H2 receptor activation leads to activation of cAMP-dependent and -independent pathways (adenylate cyclase, c-Fos, c-Jun, PKC, p70S6K). Differential levels of cellular expression result in the stimulation of hydrochloric acid secretion from acid secreting parietal cells of the gastric mucosa, smooth muscle relaxation of the vasculature and airways, increased cardiac rate and contractility, and immunomodulatory effects through basophil suppression (Novak et al. 2012). H2 receptors also mediate suppression of Th1 and Th2 responses (Jutel et al. 2001). Moreover, H2 receptors play a central role in intestinal immune homeostasis by modifying innate immune responses to bacterial components. H2 receptors mediate invariant natural killer T cell (iNKT) dependent lung inflammation in a mouse model of asthma; consequently, H2 receptor null mice show iNKT dependent lung inflammation (Ferstl et al. 2017). Comparison of H2 receptor gene expression among healthy individuals and patients with inflammatory bowel disease suggest the possibility of down regulated innate immune responses by H2 receptor signaling in the intestinal tract (Smolinska et al. 2016).
The H3 receptor is predominantly expressed in the CNS (basal ganglia, hippocampus and cortical areas), but can also be found in the peripheral nervous system, airways, the cardiovascular system, the gastrointestinal tract and on mast cells. Several isoforms are linked to different signaling pathways (Ishikawa et al. 2010) which may suggest selective regulation of diverse cerebral functions. (García-Gálvez et al. 2018). Acting through the presynaptic H3 receptor, histamine regulates its own release (negative feedback) as well as the release of other neurotransmitters such as noradrenaline, dopamine, serotonin, acetylcholine, and gamma-amino-butyric acid. In the lower airways, H3 receptors are located on postganglionic cholinergic nerves and counteracts bronchoconstriction, through stimulation of phospholipase A2 and subsequent enhanced release of endothelium-derived relaxing factor in guinea pigs (Barnes 1991; Burgaud and Oudart 1993; Nieto-Alamilla et al. 2016). In the upper airways, histamine may play a role in nasal congestion through its activity at H3 receptors. The intracellular pathways related to H3 receptor activation are enhanced Ca2+ influx, MAP kinase, and inhibition of cAMP.
The H4 receptor shows the highest level of expression in bone marrow and peripheral blood leukocytes but is also found in spleen, thymus, lung, gastrointestinal tract, liver, peripheral nerves, and central neurones in the cerebellum and hippocampus (Cogé et al. 2001). The H4 receptor is predominantly detected on hematopoietic cells, including mast cells, eosinophils, basophils, dendritic cells, monocytes, NK, and T cells (Th17). H4 receptor activation induces calcium mobilisation through cAMP in mast cells and promotes mast cells migration towards histamine. Moreover, the receptor plays a significant role in regulating dendritic and T cell function. The H4 receptor has also been implicated in the regulation of other non-hematopoietic systems such as the dermis and epidermis (resulting in stronger expression), especially in keratinocytes, intra-articular synovial cells, and chondrocytes.
The H4 receptor has been considered a promising therapeutic target in atopic dermatitis, asthma, and chronic arthritis. This relates not only to its direct activating function of effector cells of the hematopoietic system, but also to its expression on regulatory T cells. H4 receptor agonists have been shown to suppress CCL2 mRNA expression in monocyte-derived Langerhans cells, and induce the migration of Langerhans cells from the epidermis (Gschwandtner et al. 2010). The application of H4 receptor antagonists in studies showed significant inhibition of CCL17 and CCL22 production by monocytes of patients with atopic dermatitis, with only marginal effects in healthy individuals (Miyano et al. 2016). Since the application of H1 and H4 receptor antagonists decreases pruritus in atopic dermatitis, the H4 receptor has also been linked to pruritus development (Ohsawa and Hirasawa 2012; Köchling et al. 2017).
Pharmacological blockade of histamine function via antihistamines
Antihistamines were synthesized as blocking compounds against the actions of histamine; they stabilize the inactive conformation of the heptahelical H1 receptors via cross-linkage. Due to differences in their chemical structures, the site that antihistamines occupy on the H1 receptor is different from the histamine binding site, therefore, antihistamines are classified as inverse agonists rather than competitive antagonists (Church 2011) (Figure 1). Both histamine and antihistamines act by receptor stabilization, either in the active or inactive state, shifting the natural balance of the receptor state (Table 2).
Figure 1:
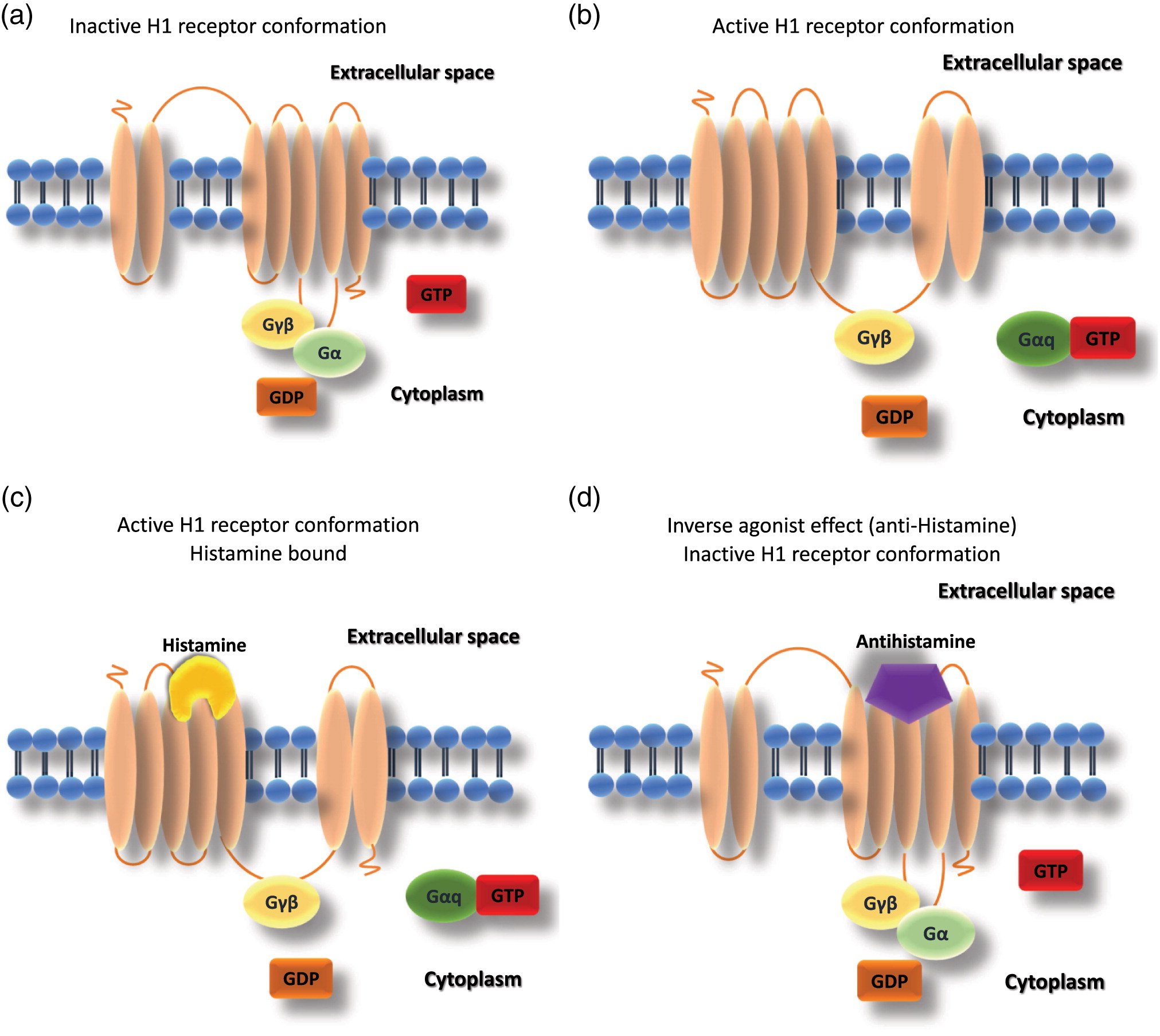
Table 2:
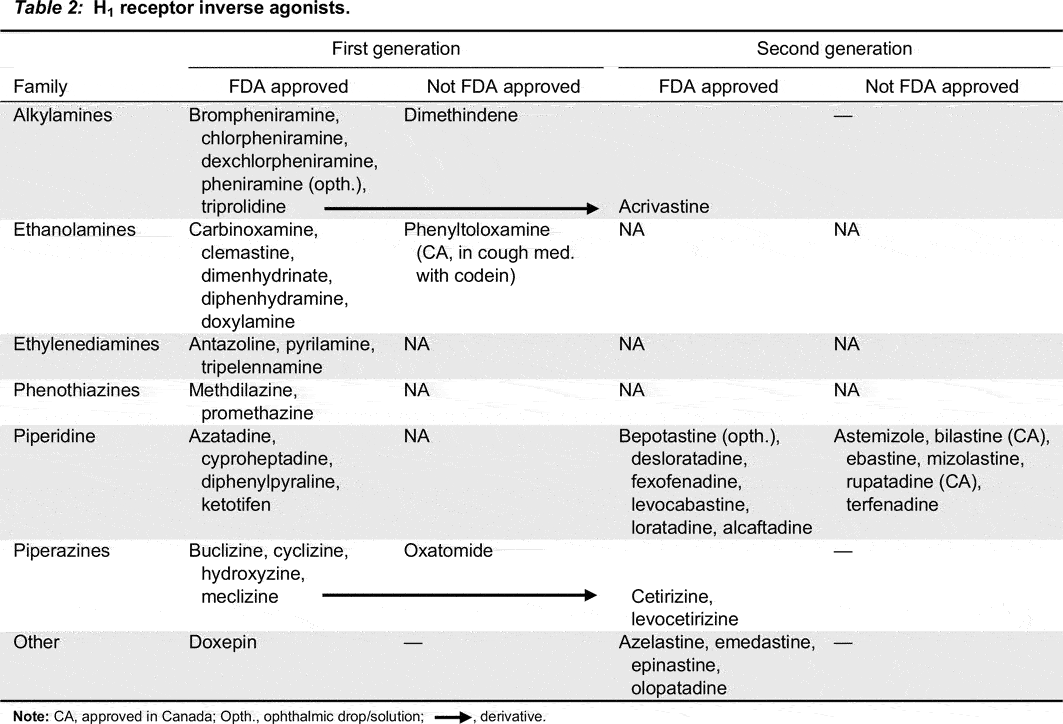
Note: CA, approved in Canada; Opth., ophthalmic drop/solution;, derivative.
Pharmacokinetics and side effects of approved antihistamines
The most important cause of side effects of first generation H1 antihistamines relates to their ability to cross the blood-brain barrier. They have low selectivity and lipophilic properties because of their basic structure of either aromatic or heterocyclic rings. This leads to decreased hepatic degradation and increase hepatobiliary circulation. Newer generation antihistamines express high H1 receptor selectivity, leading to less or no cholinergic, adrenergic or serotoninergic side effects (Church 2011; Simons and Akdis 2013).
Bioavailability
Following oral administration, antihistamines reach their peak concentration within 0.5–3 hours. Bioavailability of antihistamines is regulated by the abundance of drug transporters in the intestine. P-glycoprotein expression (P-gp) is a drug efflux pump which is expressed in a polarized fashion and found on epithelial and mucosal cells (such as the luminal surface of the intestine), on cerebral endothelial cells of the blood-brain barrier, the liver, and the kidneys. Rodent derived data suggests that antihistamines, which act as substrates of the P-gp transporter, display little to no CNS effects. This may relate to the fact that P-gp actively pumps substrates out of the brain, thereby limiting their effects in the CNS. The importance of P-gp in this context has been confirmed in particular for second generation antihistamines. P-gp−/− mice showed significantly greater enrichment of nonsedative second generation antihistamines (such as cetirizine, loratadine, and desloratadine) in the brain, as compared to wild type mice. On the other hand, brain penetration of the sedative first generation antihistamines hydroxyzine, diphenhydramine, and triprolidine did not differ between wild type and P-gp−/− mice (Chen et al. 2003; Polli et al. 2003). This apparent difference between second and first generation antihistamines is in keeping with the knowledge that only the former are P-gp substrates. P-gp inhibition and induction has the potential to disrupt antihistamine absorption and elimination. Co-administration of P-gp inhibitors such as amiodarone, propranolol, ketoconazole, and cyclosporine, carries the potential risk of increasing antihistamine exposure due to inhibition of P-gp activity in the gastrointestinal tract (Shimizu et al. 2006). Some second generation antihistamines (for example, loratadine and rupatadine) are extensively metabolized by cytochrome P450 in the liver. Thus, cytochrome P450 inhibitors, such as macrolide antibiotics and imidazole antifungals (ketoconazole), may increase the plasma concentrations of antihistamines, especially in the elderly (Kaliner 2002; del Cuvillo et al. 2006). There is a substantial overlap between drugs that interfere with cytochrome P450 3A4 (CYP3A4) and P-gp. Importantly, dual inhibitors of P-gp and CYP3A4 do not necessarily have the same inhibitory potency towards P-gp and CYP3A4. Potential drug interactions should always be evaluated in the context of characteristics of both the individual drug and patient. Potential drug-drug interactions may be handled by additional monitoring, dose adjustment, staggered administration, paused treatment and search for alternative treatments.
Selectivity
Antihistamines have organ-receptor specific affinity and efficacy. Depending on the receptor location and the histamine/antihistamine action, site effects can be organ specific. Consequently, blockade of the central nervous H1 receptor leads to somnolence, fatigue, increased appetite, disturbed circadian sleep-wake cycle and altered rapid eye movement sleep, decreased cognitive functions (impaired memory and learning) and may lead to aggressive behaviour and seizures. The interaction between H1 blockers and the H1 receptor in the brain was directly demonstrated by positron-emission tomography studies. Antihistamines can be classified based on these studies of receptor occupancy: Sedative, where receptor CNS occupancy is 50%–100%; less-sedative, where receptor occupancy is 20%–50%; and non-sedative, where receptor occupancy is 0%–20%. The limitations of these studies is the different and limited number of time points receptor occupancy analysis was performed, as well as the lack of quantitative dose/serum concentration/Tmax in relation to the receptor occupancy (Kanamitsu et al. 2017). To assess treatment safety, proportional impairment ratio was implemented to describe the correlation between the brain H1 receptor occupancy and the somnolence observed. Two studies revealed that the potential of antihistamines to impair cognitive function and performance was different both between generations of antihistamines, as well as between antihistamines of the same generations (Shamsi and Hindmarch 2000; McDonald et al. 2008). In the first generation group, hydroxyzine, chlorpheniramine, ketotifen, and diphenhydramine had the highest proportional impairment ratio.
First generation oral antihistamines have no site specific receptor selectivity. Thus, in addition to peripheral and central nervous side effects they have the potential to exert anti-alpha-adrenergic, anti-serotoninergic and anti-muscarinergic side effects. These side effects may present with dry mouth, photophobia, mydriasis, tachycardia, constipation/urinary retention, agitation, confusion. This has not been reported for second generation antihistamines.
Another important aspect is the increased risk of cardiac side effects of any H1 antihistamine that prolongs the QT interval (measurement made on an electrocardiogram) in patients with pre-existing heart disease, cardiac arrhythmias, or electrolyte imbalance. During the pre-clinical and early clinical development of new drugs, regulatory agencies worldwide now aim to identify all new medications, including H1 antihistamines, that may block the rapid delayed rectifier current and thus interfere with cardiac electrical cycle repolarization (IKr current, which conducts potassium ions out of the cardiac myocytes and is critical in timing of the repolarization), prolong the QT interval, and potentially cause polymorphic ventricular arrhythmias. Second generation H1 antihistamines such as cetirizine, desloratadine, fexofenadine, and loratadine appear to be relatively free of cardiac toxic effects as compared with astemizole and terfenadine (Woosley 1996; Simons and Simons 1999; De Bruin et al. 2002; Yap and Camm 2002; Simons and Akdis 2013).
Antihistamines in atopic disease
Chronic urticaria
According to the European Academy of Allergy and Clinical Immunology (EAACI)/Global Allergy and the Asthma European Network (GA2LEN)/the European Dermatology Forum (EDF)/the World Allergy Organization (WAO) Guideline, chronic urticaria is defined as a “condition characterized by the development of wheals (hives), angioedema or both in the case of these symptoms lasting more than 6 weeks” (Dressler et al. 2018). Histamine is the central mediator in urticaria and promotes the wheal and flare reaction via an increase of vascular permeability and vasodilation. A previous GA2LEN position paper suggests not to use first generation sedating antihistamines in allergy due to side effects (Church et al. 2010). Non-sedating H1 antihistamines are recommended as baseline treatment, to be started with a standard dose and increased up to 4-fold of the recommended dose. Omalizumab is recommended as add-on treatment in the case of unresponsiveness for high dose of H1 antihistamines (Dressler et al. 2018).
Allergic rhinitis
Allergic rhinitis (AR) is defined as an IgE-mediated inflammatory response of the nasal mucosa after exposure to allergens. Symptoms involve anterior or posterior rhinorrhea, sneezing, nasal obstruction and (or) itching of the nose (Bousquet et al. 2008). Many reports indicate that managing allergic rhinitis is beneficial for better asthma control. Therefore, as a state-of-the-art review for the specialist and the general practitioner as well as other health care professionals, Bousquet et al. (2001) published the Allergic Rhinitis and its Impact on Asthma (ARIA) document. ARIA guidelines were subsequently updated in 2008 (Bousquet et al. 2008), while a 2016 revision was limited to only 6 clinical questions (Brożek et al. 2017).
The ARIA 2008 Update recommended intranasal glucocorticosteroids as the most effective drugs for adult and pediatric patients with AR. Topical H1 antihistamines and second generation oral H1 antihistamines are recommended. However, with regards to the effectiveness of second generation H1 antihistamines, first generation oral H1 antihistamines are less effective and therefore not recommended (Bousquet et al. 2008). The ARIA 2016 revision comments on combination therapies and a leukotriene receptor antagonist (LTRA) as treatment options. Either an intranasal corticosteroid may be applied or co-administered with an oral H1 antihistamine. An intranasal H1 antihistamine is available as intranasal application alone, while a combination of an intranasal H1 antihistamine and an intranasal corticosteroid is more effective than an intranasal H1 antihistamine alone. The use of an oral or intranasal H1 antihistamine depends on the patient’s preferences and local availability. For patients with persistent AR, an oral H1 antihistamine is recommended rather than a LTRA (Brożek et al. 2017).
Allergic conjunctivitis
The symptoms of environmental (seasonal and perennial) allergic conjunctivitis are caused by IgE mediated hypersensitivity and local mast cell degranulation, as well as release of inflammatory mediators and histamine. Besides allergen avoidance, both topical and oral antihistamines are the recommended treatment of choice for symptomatic management of allergic conjunctivitis, alone or in combination with mast cell stabilisers and topical anti-inflammatory medications (non-steroidal and steroidal). Topical antihistamines used alone have a short (up to 4 hours) effect, indicating frequent 4×/day use, and do not have anti-inflammatory effects, which is important in the prevention and management of chronic inflammatory changes (La Rosa et al. 2013).
Asthma
Asthma is defined as a heterogeneous disease, usually characterized by chronic airway inflammation by Global Initiative for Asthma (GINA) (GINA 2018). A definition by the British Thoracic Society (BTS)/Scottish Intercollegiate Guideline Network (SIGN) also included airway hyperresponsiveness and airway inflammation (SIGN 2016). H1 antihistamines are not referred to in either. Oral H1 antihistamines are recommended only in asthma patients with concomitant AR (Brożek et al. 2017).
Food allergy
Food allergy is defined as adverse reaction to food mediated by an immunologic mechanism, involving specific IgE (IgE-mediated), cell-mediated mechanisms (non-IgE-mediated), or both IgE- and cell-mediated mechanisms (mixed IgE- and non-IgE-mediated) by EAACI Food Allergy and Anaphylaxis Guidelines (Muraro et al. 2014). Currently, avoidance of allergenic foods and the use of rescue medication in case of an allergic reaction is still the treatment of choice (Muraro et al. 2017; Pajno et al. 2018). H1 antihistamines should be used as rescue medication in emergency cases such as anaphylaxis, but notably not initial medications (Simons et al. 2014). According to EAACI treatment guidelines, 1st-line medication for treatment of anaphylaxis is an intramuscular epinephrine injection which could be repeated within 10 minutes if indicated, 2nd-line are inhaled beta-2 agonists for wheezing and inhaled adrenaline for stridor, and 3rd-line are H1 antihistamines, H2 antihistamines, and glucocorticoids (Muraro et al. 2014).
Novel applications of antihistamines
Recent progress in identifying new H4 antagonists resulted in derivates which are structurally related to 2 chemicals: 2-aminopyrimidine and indole carboxamides. Indole carboxamide analogs of compound 1 (JNJ7777120) was first identified as a H4 receptor antagonist by Jablonowski et al. (2003) JNJ7777120 inhibited histamine-induced eosinophil shape change and upregulation of adhesion molecules (Ling et al. 2004). A combination therapy with H1 receptor antagonist (olopatadine) and H4 receptor antagonist (JNJ777120) was effective for atopic dermatitis in mice (Matsushita et al. 2012), and in a second approach scratching behavior improved due to the same combination of H1 and H4 receptor antagonist treatment in a chronic dermatitis mouse model (Ohsawa and Hirasawa 2012). Currently, approximately 47 compounds are described as H4 receptor antagonists (Tichenor et al. 2015). The expression of the H4 receptor is limited to the area of inflammatory and immune responses on bone marrow and hematopoietic cells. Thus, the function of H4 antagonists may also involve allergic disease in humans, although this has not yet been clarified (Thurmond 2015). Some H4 receptor antagonists have reached clinical trials for the treatment of asthma, atopic dermatitis, and allergic rhinitis. JNJ39758979 reduced histamine-induced pruritus despite not demonstrating reduction in wheal or flare area (Kollmeier et al. 2014). The safety and efficacy of JNJ39758979 was assessed in adult Japanese patients with moderate atopic dermatitis. Assessment of efficacy showed improvement of several pruritus assessments (Murata et al. 2015). ZPL3893787, on the other hand, has completed Phase I in healthy volunteers with encouraging safety and pharmacokinetic profile. UR-63325 has entered into clinical studies for the treatment of allergic respiratory diseases and underwent Phase II study with additional benefit compared with H1 antagonism in reducing nasal blockage (Kiss and Keseru 2014).
Frequently used non-sedating H1 antihistamines
Cetirizine and levocetirizine
Cetirizine is a racemic mixture composed of R-levocetirizine and S-dextrocetirizine, a member of the piperazine family. It is among the early second generation H1 antihistamines (SGAHs) developed to provide selective H1 receptor inhibition with little CNS binding. It has favourable pharmacological properties in that it does not undergo interconversion and therefore maintains configuration stability in the body, and has high binding to serum albumin and the lowest apparent volume of distribution (Tillement 2000). As well, ceterizine exhibits low brain uptake, low affinity for lean tissues such as the myocardium (thus low cardiotoxicity), and low/lack of sedative effects. Cetirizine has also been shown to be absorbed extensively and rapidly from the gut (Wood et al. 1987), leading to high bioavailability and rapid onset of drug action (Greisner 2004). Unlike many other SGAHs, cetirizine does not undergo hepatic metabolism to any appreciable extent, but is excreted mostly unchanged in the urine, equally well in both healthy volunteers and patients with chronic liver disease (Horsmans et al. 1993). Dose adjustment is not needed in case of abnormal liver function but is needed in renal dysfunction (Dávila et al. 2013). The lack of hepatic metabolism demonstrates a low potential for drug-drug interactions, which avoids any exaggerated pharmacological or toxicological effects with drugs that are subject to metabolism by P450 enzymes and to transmembrane transport (Boobis et al. 2009). Onset of action of wheal and flare inhibition is 1 hour and duration of time is 24 hours (Simons 2004).
With regards to safety, cetirizine has very low to absent cardiotoxic potential. Importantly, no deleterious effects in pregnancy or lactation have been reported with cetirizine. The Food and Drug Administration (FDA) has classified cetirizine as a pregnancy category B drug, i.e., without known harm to an animal foetus and no human studies available.
Although research employing subjective assessments of the CNS effects of cetirizine has produced conflicting data, studies involving objective assessments, e.g., driving and psychomotor performance tests, have unanimously demonstrated that therapeutic doses of cetirizine do not generally produce more psychomotor impairment than placebo. Similarly, the adverse effects profile of cetirizine has been shown to be comparable with that of astemizole, ebastine, fexofenadine, loratadine, mizolastine, or terfenadine; with the adverse effects of cetirizine generally being of mild-to-moderate intensity. Importantly, the longest clinical trial with any antihistamine launched in the 20th century, the ETAC study, in children as young as 18–24 months at randomization, demonstrated that continuous cetirizine treatment over 18 months had no adverse effects on neurological and behavioural events, nor on natural development milestones, e.g., no influence on height, body mass, gross and fine motor skills, speech and language skills, as well as haematology, and biochemistry tests. The proportion of treatment-emergent adverse events in the ETAC study, as described by the investigators, was similar between the cetirizine and placebo group.
Levocetirizine, is the active enantiomer of cetirizine. Levocetirizine is poorly metabolized because of the lack of CYP2D6 (Baltes et al. 2001). There is only a need for dose adjustment in the event of serious renal failure (Dávila et al. 2013). The time it takes to reach the peak plasma concentration is 0.9 hour and the terminal elimination half-life is 7.9 hours (Baltes et al. 2001). Symptoms score for allergic rhinitis improved within 2 hours after dosing (Horak et al. 2010). Based on suppression tests of wheal and flare, time of action is also 2 hours and duration of action is 24 hours (Simons 2004). Levocetirizine is a designated FDA Pregnancy Category B (animal studies negative, human data not available, or animal studies positive, human data negative).
Long-term safety of the second generation H1 antihistamines, desloratadine and loratadine, has been documented in randomized controlled trials lasting 6–18 months in adults and in children as young as 1–2 years old (Simons et al. 2003; Simons and Early Prevention of Asthma in Atopic Children (EPAAC) Study Group 2007; Klimek 2009).
Loratadine and desloratadine
Loratadine is a second generation, oral histamine H1 receptor antagonist of the piperidine family, metabolized by the hepatic cytochrome P450 system to the active, desloratadine metabolite. Desloratadine is further processed to 3-hydroxydesloratadine, which undergoes further glucuronidation in the liver. Studies have demonstrated its efficacy for seasonal allergic rhinitis (SAR) via a reduction of symptoms over placebo. Time to onset of action has been reported widely from 75 to 180 minutes in clinical situations (Simons 2004; Tenn et al. 2018) and the duration of action is 24 hours (Simons 2004). Dose adjustment is only needed in case of serious renal failure. Minimal or no adverse effects are reported with 10 mg of loratadine. At higher doses it might cause dose-related CNS effects. Up to 30-fold overdoses of loratadine have not been causally associated with serious adverse events or fatality. A study by Wu et al. (2012) comparing the efficacy of treatment for persistent allergic rhinitis (PAR) in children 2–12 years old of loratadine syrup formulation and cyproheptadine solution described a significantly reduced total symptom score with an improvement in side effects involving the central nervous system in the loratadine group. In the Preventia I study, well tolerated long term safety effects were described in the 12–30 months of age group during Phase I studies. 12 months medication administration and Phase II 12 months follow up period (Grimfeld et al. 2004).
Loratadine is a designated FDA Pregnancy Category B. Previous studies indicated loratadine as a possible factor for the increased incidence of hypospadias in pregnancy (Pedersen et al. 2006). But according to a systematic review and meta-analysis, this risk could not be detected (Schwarz et al. 2008).
Desloratadine is a metabolite of loratadine (as well as rupatadine) and is well metabolized by the hepatic cytochrome P450 system. The enzyme pathway involved in the liver metabolism of desloratadine to 3-hydroxydesloratadine has not yet been reported. It undergoes less first-pass metabolism than loratadine; therefore, the variability in systemic exposure to desloratadine is potentially decreased. Elimination occurs via both the renal and faecal pathways (Affrime et al. 2002). The bioavailability of desloratadine is minimally affected by drugs interfering with transporter molecules; of the second generation antihistamines, desloratadine has the greatest binding affinity for the H1 receptor (Anthes et al. 2002). In an in vitro study investigating inverse agonism, desloratadine appeared to be more potent than fexofenadine and cetirizine, a result explained by the high correlation between H1 receptor affinity and inverse agonist activity (Bakker et al. 2000). Peak plasma concentrations of desloratadine at recommended doses for antihistaminic activity are 10-fold lower than the concentrations at which in vitro functional antimuscarinic activities are observed. Desloratadine does not induce any clinically relevant antimuscarinic effects at therapeutic doses. No interaction with cardiac potassium channels has been reported with desloratadine. Furthermore, desloratadine does not impair cognitive or psychomotor performance or potentiate the deleterious effects of alcohol (ethanol) on psychomotor performance, at up to 9 times its standard dose, no sedating effects have been observed (Simons 1999; Kay and Quig 2001; Scharf and Berkowitz 2007). No known clinically relevant drug-drug interactions have been observed and dose adjustment is only needed in serious renal failure (Dávila et al. 2013). Onset of action is 2 hours on the wheal and flare response and the duration of action is more than 24 hours (Simons 2004; Antonijoan et al. 2017).
Desloratadine is a designated FDA Pregnancy Category C (animal studies not available or positive, human data not available). In the US, desloratadine is approved for the treatment of SAR in adults and children aged ≥2 years, and PAR and chronic idiopathic urticaria in adults and children aged ≥6 months (prescribing information).
Rupatadine is almost completely metabolised when administered orally. Two of its main metabolites, desloratadine and 3-hydroxylated desloratadine, retain antihistaminic properties which may contribute to the overall efficacy of the drug. Although rupatadine is 98%–99% bound to human plasma proteins, it is well distributed in other tissues, indicating that this high degree of binding does not cause the compound to be retained in the circulating blood, allowing it to reach its target receptors.
Rupatadine is extensively metabolised in the liver and CYP3A4 was identified as the primary isoenzyme responsible for its metabolism. Thus, rupatadine should be used with caution when administered in combination with CYP3A4 inhibitors and should be avoided (like ketoconazole, azithromycin, fluconazole, diltiazem). Caution should be taken when rupatadine is co-administered with drugs with narrow therapeutic windows, since knowledge of the effect on other drugs is limited. No major sedating effect was described (Shamizadeh et al. 2014; Potter et al. 2016). Rupatadine has no or low side effect potential on cardiac repolarization (De Bruin et al. 2002; Yap and Camm 2002; Täubel et al. 2018); however, careful surveillance and risk-minimalization is required in patients in patients with a history of QTc prolongation and (or) torsade de pointes, including congenital long QT syndromes, or a history of other cardiac arrhythmias. There has been 1 torsade de pointes reported (Fité-Mora 2009) with rupatadine during post-market use, thus it should not be recommended to be used Rupatadine should not be administered concomitantly with erythromycin, ketoconazole or grapefruit.
Rupatadine is a generally well tolerated, rapid onset, effective second-generation H1 antihistamine for the symptomatic relief of allergic rhinitis (Guadano 2004; Fantine 2008). It is known as dual blocker, since it blocks both the action of histamine as well of other inflammatory mediators, such as platelet-activating factor. It is indicated for use in allergic rhinitis and chronic idiopathic urticaria in patients aged 2 years or more. The onset of time is 2 hours and duration of action is 24 hours (Simons 2004; Antonijoan et al. 2017). The rapid absorption of rupatadine correlates with the onset of the antihistamine and PAF actions as assessed by wheal and flare inhibition, which occurs within 1–2 hours post dose.
No adequate studies on pregnant women exists, prescribing information recommends avoiding use in pregnant women. In preclinical studies, foetal toxicity (growth delay, incomplete ossification, minor skeletal findings) was reported in rats at materno-toxic dose levels only (25 and 120 mg/kg/day). In rabbits, no evidence of developmental toxicity was noted at doses up to 100 mg/kg.
In the CNS, rupatadine behaves similarly to second generation antihistamines. Rupatadine displays psychomotor impairment activity only at the highest dose (80 mg) 4× highest therapeutic dose (Barbanoj et al. 2004). No anticholinergic effects were observed in several preclinical models at doses of up to 7 mg/kg (>40 times the human expected dose) or at single doses in the range of 10–80 mg in humans (Shamizadeh et al. 2014). A recent multicenter placebo controlled study assessing the oral solution of rupatadine in children ages 2–11 years with persistent allergic rhinitis demonstrated efficacy in both nasal and non-nasal symptom reduction, and improved quality of life in comparison to placebo (Juniper et al. 1998; Potter et al. 2013; Santamaría et al. 2018).
Bilastine
Bilastine is a new H1 receptor inverse agonist, belonging to the same non sedating piperidine family as loratadine, desloratadine, and fexofenadine. In vitro studies have shown that bilastine has a high specific affinity for the H1 receptor but no or very low affinity for other tested receptors. This affinity for the H1 receptor is 3 and 6 times higher than for cetirizine and fexofenadine, respectively. The effectiveness of bilastine is comparable to that of desloratadine. Besides its H1 antagonism, bilastine has also demonstrated anti-inflammatory properties, including inhibiting the release of proinflammatory cytokines such as interleukin (IL)-4 and tumor necrosis factor-α. Furthermore, in vivo studies in rats demonstrated a significant reduction in histamine-stimulated endothelial permeability, microvascular extravasation, and inhibition of smooth muscular contraction and bronchospasm 11 times that of cetirizine. Laboratory studies also demonstrated that bilastine may reduce passive cutaneous anaphylaxis induced by homologous serum and monoclonal antibodies, along with IgG/IgE dependent active cutaneous anaphylaxis. Studies in humans have also shown its ability to rapidly inhibit the erythematous area in response to histamine.
In a comparison performed in the Vienna Challenge Chamber or with natural exposure, bilastine proved to be more effective than fexofenadine and as effective as cetirizine in subjects with grass AR (Horak et al. 2010). With regard to pharmacokinetics, in the fasting state, bilastine is absorbed quickly and does not undergo significant metabolization. Bilastine has no impact on the cytochrome P450 enzyme system of the liver, and there is no evidence of interaction with other drugs except for an increased uptake of bilastine when taken concomitantly with ketoconazole, erythromycin, or diltiazem. Approximately 95% is excreted intact in feces (67%) and urine (33%). Therefore, in case of renal and liver failure, dose adjustments is not necessary, however the use of bilastine and a concomitant drug with a pharmacokinetics through the P-gp system should be avoided in case of renal failure (Dávila et al. 2013). Bilastine presents a favorable safety profile with regard to CNS and cardiovascular effects. Its safety profile in terms of adverse effects was very similar to that of placebo in all phase I, II, and III clinical trials. Its distribution in the brain is undetectable. Bilastine (20 mg), unlike cetirizine, does not increase the effects of alcohol on the CNS, does not increase the CNS depressant effect of lorazepam, and is similar to placebo in the driving test. No prolongation of the QTc interval (even at doses higher than the therapeutic dose) has been seen. Bilastine is well tolerated at all doses in the healthy population. The incidence of side effects was similar to that of placebo and lower than that of cetirizine with regard to fatigue and somnolence. The flare and wheal inhibition occurred at 30 and 60 minutes, respectively and duration of action is more than 24 hours (Antonijoan et al. 2017).
Fexofenadine
Fexofenadine is an active metabolite of terfenadine. Fexofenadine is now widely used as an alternative drug for terfenadine which had a risk of inducing severe ventricular arrhythmia. It is very poorly metabolized by cytochrome P450 and occurs primarily in the gastrointestinal tract. Some transporters involving P-gp, organic anion transporting polypeptide (OATP) 2B1, and multidrug resistance-associated protein 2 (MRP2) contribute to the uptake of fexofenadine (Akamine and Miura 2018; Akamine et al. 2019). Fexofenadine does not require dose adjustment (Dávila et al. 2013). It’s Tmax, onset of action, and duration of action is 2.6, 2, and 24 hours, respectively (Simons 2004). Fexofenadine is a designated FDA Pregnancy Category C. Reduction in pup weight and survival were observed and human data was not available with fexofenadine (Kar et al. 2012).
Discussion
Atopic diseases are a great burden to patients, from the young to the elderly. The prevalence of these conditions are increasing and expected to further increase in the future. Increased awareness by general practitioners, both of the conditions and therapeutic options, is will help to recognise and properly treat them. Here, we provide a current update and review of possible antihistamine effects and side effects, to aid practitioners in their treatment choice.
The different histamine receptors are distributed uniquely in the human body, with specific tissue localisation explaining their various functions and effects. From the H1 receptors’ diverse distribution and various effector activity, including in allergic rhinitis, asthma, atopic dermatitis, conjunctivitis and urticaria development, through to the H2 receptors’ effect on gastric mucosa, smooth muscle relaxation of the vasculature and the airways, cardiac rate and contractility, and immunomodulatory effects by basophil suppression. As well, the various isoforms of H3 receptors which are linked to different signaling pathways leading to selective regulation of diverse cerebral functions, and the H4 receptors’ direct activating function of effector cells of the haematopoietic system but also to the expression on regulatory T cells. These are all explored in order to better understand their mechanism, effect, and role in the development of atopic diseases and developing new therapeutic agents for the management of atopic dermatitis, allergies, and asthma. The improving knowledge of receptor action at the cellular level helps to develop antihistamines with less side effects and more selectivity. Understanding the structure related side effects of first generation antihistamines, leading to sedation, helped in the development of the less sedating compounds.
Bioavailability of antihistamines is regulated by the abundance of drug transporters in the intestine. The significance of P-gp expression is in its limiting effects on the CNS, by actively pumping out the substrate, in this case, second generation non sedative antihistamines that may pass through the blood brain barrier. Thus, P-gp inhibition and induction has the potential to disrupt antihistamine absorption and elimination. Its importance is highlighted by the potential side effects of co-administration of P-gp inhibitors (e.g., amiodarone, propranolol, ketoconazole, and cyclosporine). Some second generation antihistamines, such as loratadine and rupatadine, are extensively metabolized by cytochrome P450 in the liver. Therefore, co-adminitration of CYP450 inhibitors, such as macrolide antibiotics and imidazole antifungals (ketoconazole) may increase the plasma concentrations of these antihistamines, especially in the elderly. Potential drug-drug interactions may be handled by additional monitoring, dose adjustment, staggered administration, paused treatment and search for alternative treatment.
Antihistamines have organ-receptor specific affinity and efficacy. Depending on the receptor location and the histamine/antihistamine action, site effects can be organ-specific. First generation oral antihistamines do not have site specific receptor selectivity, thus, in addition to peripheral and central nervous side effects they have the potential to exert anti-alpha-adrenergic, anti-serotoninergic, and anti-muscarinergic side effects. These important side effects present with dry mouth, photophobia, mydriasis, tachycardia, constipation/urinary retention, agitation, and confusion. This has not been reported for second generation antihistamines. Another important aspect is the increased risk of cardiac side effects of any H1 antihistamine that prolongs the QT interval in patients with pre-existing organic heart disease, cardiac arrhythmias, or electrolyte imbalance, however second-generation H1-antihistamines appear to be relatively free of cardiac toxic effects.
In the therapy of atopic diseases, like chronic urticaria, non sedating H1 antihistamines are recommended as baseline treatment, to be started with a standard dose and increased up to 4-fold of the recommended dose. In allergic rhinitis many reports indicate that intranasal glucocorticosteroids combined with topical H1 antihistamines or second generation oral H1 antihistamines is the most recommended effective drugs therapy for adult and children patients with AR. However patient preferences and co-morbidities largely affect the medication and the administration route (therapy of choice). Oral antihistamine therapy is not recommended as part of the therapeutical regimen for asthma; oral H1 antihistamines are recommended in asthma patients only in case of concomitant AR. Food Allergy and Anaphylaxis Guidelines do not recommend the use of antihistaminesas part of the initial medication choice. The first line medication of anaphylaxis is intramuscular epinephrine injection, which could be repeated if indicated in 5–10 minutes, 2nd-line are inhaled beta-2 agonists for wheezing and inhaled adrenaline for stridor, and 3rd-line are second generation H1 antihistamines, H2 antihistamines, and glucocorticoids. H1 antihistamines should be used as rescue medication in emergency case such as anaphylaxis but notably not initial medications.
In this article, we provide a review of the histamine receptor, their function and role in disease, the most important antihistamines currently available, as well as their pharmacological effects and side effect potential. This knowledge will help practitioners use antihistamines in the right context when treating atopic diseases. The general recommendation is to avoid first generation sedation antihistamines with very few exception. Second generation non sedating antihistamine are generally considered very safe medications. Nevertheless, careful assessment of potential side effects is important before starting long term or high dose treatment for each antihistamine medication including concomittant illnesses and therapies (medication, herbal remedies, OTC medications) to achieve the best individualized therapy.
REFERENCES
Affrime M., Gupta S., Banfield C., and Cohen A. 2002. A pharmacokinetic profile of desloratadine in healthy adults, including elderly. Clin. Pharmacokinet. 41:13–19.
Akamine Y. and Miura M. 2018. An update on the clinical pharmacokinetics of fexofenadine enantiomers. Expert Opin. Drug Metab. Toxicol. 14(4):429–434.
Akamine Y., Yasui-Furukori N., and Uno T. 2019. Drug-drug interactions of P-gp substrates unrelated to CYP metabolism. Curr. Drug Metab. 20(2):124–129.
Anthes J.C., Gilchrest H., Richard C., Eckel S., Hesk D., West R.E. Jr., Williams S.M., Greenfeder S., Billah M., Kreutner W., and Egan R.E. 2002. Biochemical characterization of desloratadine, a potent antagonist of the human histamine H1 receptor. Eur. J. Pharmacol. 449:229–237.
Antonijoan R., Coimbra J., García-Gea C., Puntes M., Gich I., Campo C., Valiente R., and Labeaga L. 2017. Comparative efficacy of bilastine, desloratadine and rupatadine in the suppression of wheal and flare response induced by intradermal histamine in healthy volunteers. Curr. Med. Res. Opin. 33(1):129–136.
Bakker R.A., Wieland K., Timmerman H., and Leurs R. 2000. Constitutive activity of the histamine H1 receptor reveals inverse agonism of histamine H1 receptor antagonists. Eur. J. Pharmacol. 387(1):R5–R7.
Baltes E., Coupez R., Giezek H., Voss G., Meyerhoff C., and Benedetti M.S. 2001. Absorption and disposition of levocetirizine, the eutomer of cetirizine, administered alone or as cetirizine to healthy volunteers. Fundam. Clin. Pharmacol. 15(4):269–277.
Barbanoj M.J., García-Gea C., Morte A., Izquierdo I., Pérez I., and Jané F. 2004. Central and peripheral evaluation of rupatadine, a new antihistamine/platelet-activating factor antagonist, at different doses in healthy volunteers. Neuropsychobiology. 50(4):311–321.
Barnes P.J. 1991. Histamine receptors in the lung. Agents Actions Suppl. 33:103–122.
Birdsall N.J.M. 1991. Cloning and structure-function of the H2 histamine receptor. Trends Pharmacol. Sci. 12(1):9–10.
Bockaert J. and Pin J.P. 1999. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 18(7):1723–1729.
Boobis A., Watelet J.B., Whomsley R., Benedetti M.S., Demoly P., and Tipton K. 2009. Drug interactions. Drug Metab. Rev. 41(3):486–527.
Bousquet J., van Cauwenberge P., and Khaltaev N. 2001. Allergic Rhinitis and its Impact on Asthma. J. Allergy Clin. Immunol. 108(5 Suppl.):S147–S334.
Bousquet J., Khaltaev N., Cruz A.A., Denburg J., Fokkens W.J., Togias A., Zuberbier T., Baena-Cagnani C.E., Canonica G.W., van Weel C., Agache I., Aït-Khaled N., Bachert C., Blaiss M.S., Bonini S., Boulet L.P., Bousquet P.J., Camargos P., Carlsen K.H., Chen Y., Custovic A., Dahl R., Demoly P., Douagui H., Durham S.R., van Wijk R.G., Kalayci O., Kaliner M.A., Kim Y.Y., Kowalski M.L., Kuna P., Le L.T., Lemiere C., Li J., Lockey R.F., Mavale-Manuel S., Meltzer E.O., Mohammad Y., Mullol J., Naclerio R., O’Hehir R.E., Ohta K., Ouedraogo S., Palkonen S., Papadopoulos N., Passalacqua G., Pawankar R., Popov T.A., Rabe K.F., Rosado-Pinto J., Scadding G.K., Simons F.E., Toskala E., Valovirta E., van Cauwenberge P., Wang D.Y., Wickman M., Yawn B.P., Yorgancioglu A., Yusuf O.M., Zar H., Annesi-Maesano I., Bateman E.D., Ben Kheder A., Boakye D.A., Bouchard J., Burney P., Busse W.W., Chan-Yeung M., Chavannes N.H., Chuchalin A., Dolen W.K., Emuzyte R., Grouse L., Humbert M., Jackson C., Johnston S.L., Keith P.K., Kemp J.P., Klossek J.M., Larenas-Linnemann D., Lipworth B., Malo J.L., Marshall G.D., Naspitz C., Nekam K., Niggemann B., Nizankowska-Mogilnicka E., Okamoto Y., Orru M.P., Potter P., Price D., Stoloff S.W., Vandenplas O., Viegi G., and Williams D. 2008. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008. Allergy. 63(Suppl. 86):8–160.
Brożek J.L., Bousquet J., Agache I., Agarwal A., Bachert C., Bosnic-Anticevich S., Brignardello-Petersen R., Canonica G.W., Casale T., Chavannes N.H., Correia de Sousa J., Cruz A.A., Cuello-Garcia C.A., Demoly P., Dykewicz M., Etxeandia-Ikobaltzeta I., Florez I.D., Fokkens W., Fonseca J., Hellings P.W., Klimek L., Kowalski S., Kuna P., Laisaar K.T., Larenas-Linnemann D.E., Lødrup Carlsen K.C., Manning P.J., Meltzer E., Mullol J., Muraro A., O’Hehir R., Ohta K., Panzner P., Papadopoulos N., Park H.S., Passalacqua G., Pawankar R., Price D., Riva J.J., Roldán Y., Ryan D., Sadeghirad B., Samolinski B., Schmid-Grendelmeier P., Sheikh A., Togias A., Valero A., Valiulis A., Valovirta E., Ventresca M., Wallace D., Waserman S., Wickman M., Wiercioch W., Yepes-Nuñez J.J., Zhang L., Zhang Y., Zidarn M., Zuberbier T., and Schünemann H.J. 2017. Allergic Rhinitis and its Impact on Asthma (ARIA) guidelines—2016 revision. J. Allergy Clin. Immunol. 140(4):950–958.
Burgaud J.L. and Oudart N. 1993. Bronchodilatation of guinea-pig perfused bronchioles induced by the H3-receptor for histamine: Role of epithelium. Br. J. Pharmacol. 109(4):960–966.
Chen C., Hanson E., Watson J.W., and Lee J.S. 2003. P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Metab. Dispos. 31(3):312–318.
Church M.K. 2011. Comparative inhibition by bilastine and cetirizine of histamine-induced wheal and flare responses in humans. Inflamm. Res. 60(12):1107–1112.
Church M.K., Maurer M., Simons F.E.R., Bindslev-Jensen C., van Cauwenberge P., Bousquet J., Holgate S.T., and Zuberbier T. 2010. Risk of first-generation H1-antihistamines: A GA2LEN position paper. Allergy. 65(4):459–466.
Cogé F., Guénin S.P., Rique H., Boutin J.A., and Galizzi J.P. 2001. Structure and expression of the human histamine H4-receptor gene. Biochem. Biophys. Res. Commun. 284(2):301–309.
Dávila I., del Cuvillo A., Mullol J., Jáuregui I., Bartra J., Ferrer M., Montoro J., Sastre J., and Valero A. 2013. Use of second generation H1 antihistamines in special situations. J. Investig. Allergol. Clin. Immunol. 23(Suppl. 1):1–16.
De Bruin M.L., van Puijenbroek E.P., Egberts A.C.G., Hoes A.W., and Leufkens H.G.M. 2002. Non-sedating antihistamine drugs and cardiac arrhythmias—Biased risk estimates from spontaneous reporting systems? Br. J. Clin. Pharmacol. 53(4):370–374.
del Cuvillo A., Mullol J., Bartra J., Dávila I., Jáuregui I., Montoro J., Sastre J., and Valero A.L. 2006. Comparative pharmacology of the H1 antihistamines. J. Investig. Allergol. Clin. Immunol. 16(Suppl. 1):3–12.
Dressler C., Rosumeck S., Werner R.N., Magerl M., Metz M., Maurer M., Nast A., and Zuberbier T. 2018. Executive summary of the methods report for ‘The EAACI/GA2LEN/EDF/WAO guideline for the definition, classification, diagnosis and management of urticaria. The 2017 revision and update’. Allergy. 73(5):1145–1146.
Ferstl R., Frei R., Barcik W., Schiavi E., Wanke K., Ziegler M., Rodriguez-Perez N., Groeger D., Konieczna P., Zeiter S., Nehrbass D., Lauener R., Akdis C.A., and O’Mahony L. 2017. Histamine receptor 2 modifies iNKT cell activity within the inflamed lung. Allergy. 72(12):1925–1935.
Fité-Mora R. 2009. Torsade de pointes relacionadas con el uso de rupatadina. Rev. Esp. Cardiol. 62(3):330–331; author reply 332.
García-Gálvez A.-M., Escamilla-Sanchez J., Flores-Maldonado C., Contreras R.-G., Arias J.-M., and Arias-Montano J.-A. 2018. Differential homologous desensitization of the human histamine H3 receptors of 445 and 365 amino acids expressed in CHO-K1 cells. Neurochem. Int. 112:114–123.
Greisner W.A. 2004. Onset of action for the relief of allergic rhinitis symptoms with second-generation antihistamines. Allergy Asthma Proc. 25(2):81–83.
Grimfeld A., Holgate S.T., Canonica G.W., Bonini S., Borres M.P., Adam D., Canseco Gonzalez C., Lobaton P., Patel P., Szczeklik A., Danzig M.R., Roman I., Bismut H., and Czarlewski W. 2004. Prophylactic management of children at risk for recurrent upper respiratory infections: The Preventia I Study. Clin. Exp. Allergy. 34(11):1665–1672.
Horak F., Zieglmayer P., Zieglmayer R., and Lemell P. 2010. The effects of bilastine compared with cetirizine, fexofenadine, and placebo on allergen-induced nasal and ocular symptoms in patients exposed to aeroallergen in the Vienna Challenge Chamber. Inflamm. Res. 59(5):391–398.
Horsmans Y., Desager J.P., Hulhoven R., and Harvengt C. 1993. Single-dose pharmacokinetics of cetirizine in patients with chronic liver disease. J. Clin. Pharmacol. 33(10):929–932.
Ishikawa M., Watanabe T., Kudo T., Yokoyama F., Yamauchi M., Kato K., Kakui N., and Sato Y. 2010. Investigation of the histamine H3 receptor binding site. Design and synthesis of hybrid agonists with a lipophilic side chain. J. Med. Chem. 53(17):6445–6456.
Jablonowski J.A., Grice C.A., Chai W., Dvorak C.A., Venable J.D., Kwok A.K., Ly K.S., Wei J., Baker S.M., Desai P.J., Jiang W., Wilson S.J., Thurmond R.L., Karlsson L., Edwards J.P., Lovenberg T.W., and Carruthers N.I. 2003. The first potent and selective non-imidazole human histamine H4 receptor antagonists. J. Med. Chem. 46(19):3957–3960.
Juniper E.F., Howland W.C., Roberts N.B., Thompson A.K., and King D.R. 1998. Measuring quality of life in children with rhinoconjunctivitis. J. Allergy Clin. Immunol. 101(2 Pt 1):163–170.
Jutel M., Watanabe T., Klunker S., Akdis M., Thomet O.A., Malolepszy J., Zak-Nejmark T., Koga R., Kobayashi T., Blaser K., and Akdis C.A. 2001. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. 413(6854):420–425.
Kaliner M.A. 2002. H1-antihistamines in the elderly. Clin. Allergy Immunol. 17:465–481.
Kanamitsu K., Nozaki Y., Nagaya Y., Sugiyama Y., and Kusuhara H. 2017. Corrigendum to “Quantitative prediction of histamine H1 receptor occupancy by the sedative and non-sedative antagonists in the human central nervous system based on systemic exposure and preclinical data” [Drug Metab Pharmacokinet 32 (2017) 135–144]. Drug Metab. Pharmacokinet. 32(4):228.
Kar S., Krishnan A., Preetha K., and Mohankar A. 2012. A review of antihistamines used during pregnancy. J. Pharmacol. Pharmacother. 3(2):105–108.
Kay G.G. and Quig M.E. 2001. Impact of sedating antihistamines on safety and productivity. Allergy Asthma Proc. 22(5):281–283.
Kiss R. and Keseru G.M. 2014. Novel histamine H4 receptor ligands and their potential therapeutic applications: An update. Expert Opin. Ther. Pat. 24(11):1185–1197.
Klimek L. 2009. Levocetirizine: From scientific evidence to a potent modern-day treatment of today’s allergic patients. Drugs Today. 45(3):213–225.
Köchling H., Schaper K., Wilzopolski J., Gutzmer R., Werfel T., Bäumer W., Kietzmann M., and Rossbach K. 2017. Combined treatment with H1 and H4 receptor antagonists reduces inflammation in a mouse model of atopic dermatitis. J. Dermatol. Sci. 87(2):130–137.
Kollmeier A., Francke K., Chen B., Dunford P.J., Greenspan A.J., Xia Y., Xu X.L., Zhou B., and Thurmond R.L. 2014. The histamine H4 receptor antagonist, JNJ 39758979, is effective in reducing histamine-induced pruritus in a randomized clinical study in healthy subjects. J. Pharmacol. Exp. Ther. 350(1):181–187.
La Rosa M., Lionetti E., Reibaldi M., Russo A., Longo A., Leonardi S., Tomarchio S., Avitabile T., and Reibaldi A. 2013. Allergic conjunctivitis: A comprehensive review of the literature. Ital. J. Pediatr. 39(1):18.
Ling P., Ngo K., Nguyen S., Thurmond R.L., Edwards J.P., Karlsson L., and Fung-Leung W.-P. 2004. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br. J. Pharmacol. 142(1):161–171.
Liu C., Wilson S.J., Kuei C., and Lovenberg T.W. 2001. Comparison of human, mouse, rat, and guinea pig histamine H4 receptors reveals substantial pharmacological species variation. J. Pharmacol. Exp. Ther. 299(1):121–130.
Matsushita A., Seike M., Okawa H., Kadawaki Y., and Ohtsu H. 2012. Advantages of histamine H4 receptor antagonist usage with H1 receptor antagonist for the treatment of murine allergic contact dermatitis. Exp. Dermatol. 21(9):714–715.
McDonald K., Trick L., and Boyle J. 2008. Sedation and antihistamines: An update. Review of inter-drug differences using proportional impairment ratios. Hum. Psychopharmacol. 23(7):555–570.
Miyano K., Matsushita S., Tsuchida T., and Nakamura K. 2016. Inhibitory effect of a histamine 4 receptor antagonist on CCL17 and CCL22 production by monocyte-derived Langerhans cells in patients with atopic dermatitis. J. Dermatol. 43(9):1024–1029.
Muraro A., Werfel T., Hoffmann-Sommergruber K., Roberts G., Beyer K., Bindslev-Jensen C., Cardona V., Dubois A., duToit G., Eigenmann P., Fernandez Rivas M., Halken S., Hickstein L., Høst A., Knol E., Lack G., Marchisotto M.J., Niggemann B., Nwaru B.I., Papadopoulos N.G., Poulsen L.K., Santos A.F., Skypala I., Schoepfer A., Van Ree R., Venter C., Worm M., Vlieg-Boerstra B., Panesar S., de Silva D., Soares-Weiser K., Sheikh A., Ballmer-Weber B.K., Nilsson C., de Jong N.W., Akdis C.A., and EAACI Food Allergy and Anaphylaxis Guidelines Group. 2014. EAACI food allergy and anaphylaxis guidelines: Diagnosis and management of food allergy. Allergy. 69(8):1008–1025.
Muraro A., Lemanske R.F. Jr., Castells M., Torres M.J., Khan D., Simon H.U., Bindslev-Jensen C., Burks W., Poulsen L.K., Sampson H.A., Worm M., and Nadeau K.C. 2017. Precision medicine in allergic disease-food allergy, drug allergy, and anaphylaxis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma and Immunology. Allergy. 72(7):1006–1021.
Murata Y., Song M., Kikuchi H., Hisamichi K., Xu X.L., Greenspan A., Kato M., Chiou C.F., Kato T., Guzzo C., Thurmond R.L., Ohtsuki M., and Furue M. 2015. Phase 2a, randomized, double-blind, placebo-controlled, multicenter, parallel-group study of a H4R-antagonist (JNJ-39758979) in Japanese adults with moderate atopic dermatitis. J. Dermatol. 42(2):129–139.
Nakamura T., Itadani H., Hidaka Y., Ohta M., and Tanaka K. 2000. Molecular cloning and characterization of a new human histamine receptor, HH4R. Biochem. Biophys. Res. Commun. 279(2):615–620.
Nieto-Alamilla G., Márquez-Gómez R., García-Gálvez A.M., Morales-Figueroa G.E., and Arias-Montaño J.A. 2016. The histamine H3 receptor: Structure, pharmacology, and function. Mol. Pharmacol. 90(5):649–673.
Novak N., Mete N., Bussmann C., Maintz L., Bieber T., Akdis M., Zumkehr J., Jutel M., and Akdis C. 2012. Early suppression of basophil activation during allergen-specific immunotherapy by histamine receptor 2. J. Allergy Clin. Immunol. 130(5):1153–1158.e2.
Ohsawa Y. and Hirasawa N. 2012. The antagonism of histamine H1 and H4 receptors ameliorates chronic allergic dermatitis via anti-pruritic and anti-inflammatory effects in NC/Nga mice. Allergy. 67(8):1014–1022.
Pajno G.B., Fernandez-Rivas M., Arasi S., Roberts G., Akdis C.A., Alvaro-Lozano M., Beyer K., Bindslev-Jensen C., Burks W., Ebisawa M., Eigenmann P., Knol E., Nadeau K.C., Poulsen L.K., van Ree R., Santos A.F., du Toit G., Dhami S., Nurmatov U., Boloh Y., Makela M., O’Mahony L., Papadopoulos N., Sackesen C., Agache I., Angier E., Halken S., Jutel M., Lau S., Pfaar O., Ryan D., Sturm G., Varga E.M., van Wijk R.G., Sheikh A., Muraro A., and EAACI Allergen Immunotherapy Guidelines Group. 2018. EAACI guidelines on allergen immunotherapy: IgE-mediated food allergy. Allergy. 73(4):799–815.
Pedersen L., Nørgaard M., Skriver M.V., Olsen J., and Sørensen H.T. 2006. Prenatal exposure to loratadine in children with hypospadias: A nested case-control study within the Danish National Birth Cohort. Am. J. Ther. 13(4):320–324.
Polli J.W., Baughman T.M., Humphreys J.E., Jordan K.H., Mote A.L., Salisbury J.A., Tippin T.K., and Serabjit-Singh C.J. 2003. P-glycoprotein influences the brain concentrations of cetirizine (Zyrtec), a second-generation non-sedating antihistamine. J. Pharm. Sci. 92(10):2082–2089.
Potter P., Maspero J.F., Vermeulen J., Barkai L., Németh I., Baillieau R.A., Garde J.M., Giralt J., Doménech A., Izquierdo I., and Nieto A. 2013. Rupatadine oral solution in children with persistent allergic rhinitis: A randomized, double-blind, placebo-controlled study. Pediatr. Allergy Immunol. 24(2):144–150.
Potter P., Mitha E., Barkai L., Mezei G., Santamaría E., Izquierdo I., and Maurer M. 2016. Rupatadine is effective in the treatment of chronic spontaneous urticaria in children aged 2–11 years. Pediatr. Allergy Immunol. 27(1):55–61.
Santamaría E., Izquierdo I., Valle M., Vermeulen J.H., and Potter P. 2018. Rupatadine oral solution for 2–5-year-old children with allergic rhinitis: A safety, open-label, prospective study. J. Asthma Allergy. 11:225–231.
Scharf M. and Berkowitz D. 2007. Effects of desloratadine and alcohol coadministration on psychomotor performance. Curr. Med. Res. Opin. 23(2):313–321.
Schwarz E.B., Moretti M.E., Nayak S., and Koren G. 2008. Risk of hypospadias in offspring of women using loratadine during pregnancy: A systematic review and meta-analysis. Drug Saf. 31(9):775–788.
Shamizadeh S., Brockow K., and Ring J. 2014. Rupatadine: Efficacy and safety of a non-sedating antihistamine with PAF-antagonist effects. Allergo J. Int. 23:87–95.
Shamsi Z. and Hindmarch I. 2000. Sedation and antihistamines: A review of inter-drug differences using proportional impairment ratios. Hum. Psychopharmacol. 15(S1):S3–S30.
Shimizu M., Uno T., Sugawara K., and Tateishi T. 2006. Effects of itraconazole and diltiazem on the pharmacokinetics of fexofenadine, a substrate of P-glycoprotein. Br. J. Clin. Pharmacol. 61(5):538–544.
Simons F.E.R. 1999. Prospective, long-term safety evaluation of the H1-receptor antagonist cetirizine in very young children with atopic dermatitis. J. Allergy Clin. Immunol. 104(2 Pt 1):433–440.
Simons F.E.R. 2004. Advances in H1-antihistamines. N. Engl. J. Med. 351(21):2203–2217.
Simons, F.E.R., and Akdis, A.C. 2013. Histamine and H1-antihistamines. In Middleton’s allergy: Principles and practice. Edited by N.F. Adkinson, Jr., B.S. Bochner, W.W. Busse, S.T. Holgate, R.F. Lemanske, Jr., and F.E.R. Simons. St. Louis, MO, USA: Mosby. pp. 1503–1533.
Simons F.E.R. and Early Prevention of Asthma in Atopic Children (EPAAC) Study Group. 2007. Safety of levocetirizine treatment in young atopic children: An 18-month study. Pediatr. Allergy Immunol. 18(6):535–542.
Simons F.E.R. and Simons K.J. 1999. Clinical pharmacology of new histamine H1 receptor antagonists. Clin. Pharmacokinet. 36(5):329–352.
Simons F.E.R., Silas P., Portnoy J.M., Catuogno J., Chapman D., and Olufade A.O. 2003. Safety of cetirizine in infants 6 to 11 months of age: A randomized, double-blind, placebo-controlled study. J. Allergy Clin. Immunol. 111(6):1244–1248.
Simons F.E.R., Ardusso L.R., Bilò M.B., Cardona V., Ebisawa M., El-Gamal Y.M., Lieberman P., Lockey R.F., Muraro A., Roberts G., Sanchez-Borges M., Sheikh A., Shek L.P., Wallace D.V., and Worm M. 2014. International consensus on (ICON) anaphylaxis. World Allergy Organ. J. 7(1):9.
Smolinska S., Groeger D., Perez N.R., Schiavi E., Ferstl R., Frei R., Konieczna P., Akdis C.A., Jutel M., and O’Mahony L. 2016. Histamine receptor 2 is required to suppress innate immune responses to bacterial ligands in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 22(7):1575–1586.
Täubel J., Ferber G., Fernandes S., Santamaría E., and Izquierdo I. 2018. Cardiac safety of rupatadine in a single-ascending-dose and multiple-ascending-dose study in healthy Japanese subjects, using intensive electrocardiogram assessments-comparison with the previous white Caucasian thorough QT study. Clin. Pharmacol. Drug Dev. 7(1):67–76.
Tenn M.W., Steacy L.M., Ng C.C., and Ellis A.K. 2018. Onset of action for loratadine tablets for the symptomatic control of seasonal allergic rhinitis in adults challenged with ragweed pollen in the environmental exposure unit: A post hoc analysis of total symptom score. Allergy Asthma Clin. Immunol. 14(1):5.
Thurmond R.L. 2015. The histamine H4 receptor: From orphan to the clinic. Front. Pharmacol. 6:65.
Tichenor M.S., Thurmond R.L., Venable J.D., and Savall B.M. 2015. Functional profiling of 2-aminopyrimidine histamine H4 receptor modulators. J. Med. Chem. 58(18):7119–7127.
Tillement J.-P. 2000. The advantages for an H1 antihistamine of a low volume of distribution. Allergy. 55(Suppl. 60):17–21.
Timmerman H. 1992. Cloning of the H1 histamine receptor. Trends Pharmacol. Sci. 13(1):6–7.
Wood S.G., John B.A., Chasseaud L.F., Yeh J., and Chung M. 1987. The metabolism and pharmacokinetics of 14C-cetirizine in humans. Ann. Allergy. 59(6 Pt 2):31–34.
Woosley R.L. 1996. Cardiac actions of antihistamines. Annu. Rev. Pharmacol. Toxicol. 36(1):233–252.
Wu K.G., Li T.H., Wang T.Y., Hsu C.L., and Chen C.J. 2012. A comparative study of loratadine syrup and cyproheptadine HCL solution for treating perennial allergic rhinitis in Taiwanese children aged 2–12 years. Int. J. Immunopathol. Pharmacol. 25(1):231–237.
Yamashita M., Fukui H., Sugama K., Horio Y., Ito S., Mizuguchi H., and Wada H. 1991. Expression cloning of a cDNA encoding the bovine histamine H1 receptor. Proc. Natl. Acad. Sci. USA. 88(24):11515–11519.
Yap Y.G. and Camm A.J. 2002. Potential cardiac toxicity of H1-antihistamines. Clin. Allergy Immunol. 17:389–419.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 2 • June 2019
Pages: 35 - 51
History
Received: 11 December 2018
Accepted: 8 January 2019
Accepted manuscript online: 24 April 2019
Copyright
© 2019.
Authors
Funding Information
This review did not receive any specific grant or funding.
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AmarillaMandola, AsakoNozawa, and ThomasEiwegger. 2019. Histamine, histamine receptors, and anti-histamines in the context of allergic responses. LymphoSign Journal.
6(2): 35-51. https://doi.org/10.14785/lymphosign-2018-0016
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
