Report of the National Immunoglobulin Replacement Expert Committee: algorithm for diagnosis of immunodeficiency requiring antibody replacement therapy
Abstract
Immunoglobulin replacement therapy is a mainstay in the treatment of immune deficiencies characterized by antibody failure. Whether the cause is primary or secondary, affected patients frequently present with a history recurrent and complicated infections of the upper and (or) lower respiratory tract. Such replacement therapy has been available since the 1980s, although treatment modalities have since been refined to provide improved protection against infections resulting in reduced morbidity and mortality. Here, we describe an algorithm for diagnosing patients with suspected primary or secondary immunodeficiency, including assessment of clinical, laboratory, and genetic information, when considering initiating immunoglobulin replacement. The increasing availability of molecular genetic techniques will likely result in decreased diagnostic delay for these patients.
Statement of novelty: We describe here an algorithm for diagnosing patients with immunodeficiency requiring immunoglobulin replacement therapy.
Immunoglobulin replacement therapy is a mainstay in the treatment of humoral immune deficiencies, including many well characterized primary immunodeficiencies (PID) (Picard et al. 2015) as well as secondary immunodeficiencies (SID). The widespread availability of intravenously infused immunoglobulin (IVIG) from the 1980s opened the door towards more effective therapeutic options for those with antibody failure, by normalizing serum IgG levels and providing improved outcomes for infections. Early studies by Roifman and colleagues demonstrated an improvement in lung function in patients receiving a higher dose of IVIG, 600 mg/kg, compared to a lower dose of 200 mg/kg (Roifman et al. 1987). In the decades since, indices of efficacious immunoglobulin replacement therapy have been refined, including a move to more individualized IgG trough levels (ranging from 500 to 1700 mg/dL) that prevent recurrent and complicated infections (Lucas et al. 2010).
While the underlying cause of PID and SID differ, the clinical manifestations are usually similar, most commonly, infections of the upper and (or) lower respiratory tract caused by encapsulated bacteria as well as infections of the gastrointestinal tract. Here, we describe the algorithm for diagnosing patients with suspected PID or SID, including assessment of clinical, laboratory, and genetic information, as well as considerations for initiation of immunoglobulin replacement therapy.
Clinical suspicion of PID (including severe combined immunodeficiency, SCID; combined immunodeficiency, CID; common variable immunodeficiency disorder, CVID; or unclassified primary antibody deficiencies) and SID (due to B cell lymphoproliferative disease, protein loss, abnormal lymphatic circulation, drug-related therapies, or increased catabolism) requires thorough patient history assessment and laboratory work up (Figure 1).
Figure 1:
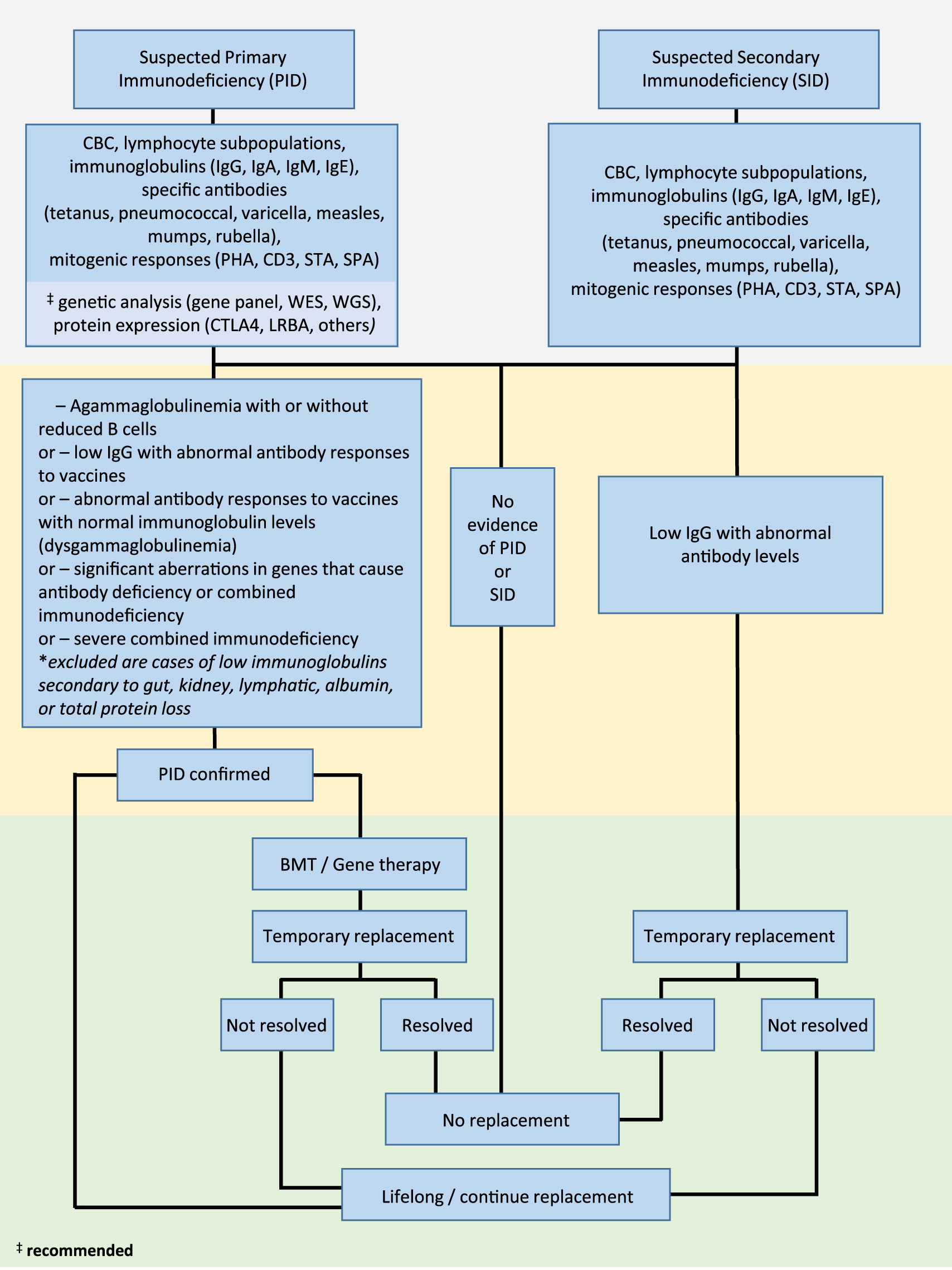
Given that antibody deficiency is characterized by recurrent infections, laboratory parameters that assess baseline and functional immune responses are essential. These include a complete blood count (CBC), flow cytometric determination of lymphocyte phenotypes (T, B, and NK cell types), serum levels of immunoglobulins (IgG, IgA, IgM, and IgE), and determination of specific antibody responses to vaccines (both protein vaccines, such as tetanus toxoid, as well as carbohydrate type vaccines, including polysaccharide pneumococcal). Assessment of lymphocyte proliferation to mitogens (phytohemagglutinin, CD3, STA, and SP3) should be performed for suspected PID. Where possible, genetic analysis (assessment by panel of known genes involved in immunodeficiency, or next generation sequencing techniques including whole exome or whole genome sequencing) can be applied to identify disease-causing variants. Similarly, flow cytometric determination of suspected cell anomalies can also be performed.
Where PID is confirmed with findings of agammaglobulinemia with or without reduced B cells, or low IgG with abnormal antibody responses to vaccines, abnormal antibody responses to vaccines with normal immunoglobulin levels (dysgammaglobulinemia), significant aberrations in genes that cause antibody deficiency, or diagnosis of SCID, immunoglobulin replacement therapy should be initiated. Where appropriate, considerations should be made for curative bone marrow transplantation or gene therapy.
In cases where SID is confirmed with evidence of low IgG in combination with abnormal antibody levels, immunoglobulin replacement should be initiated on a temporary basis until the antibody failure has resolved.
In summary, the decision to proceed with immunoglobulin replacement should be made following thorough assessment of clinical and laboratory findings. It is important to note that the true efficacy of replacement therapy should be based on the prevention of infections, rather than serum IgG levels, leading to reduced morbidity and mortality and improved quality of life. The increasing availability of molecular genetic techniques will likely result in decreased diagnostic delay for these patients.
REFERENCES
Lucas M., Lee M., Lortan J., Lopez-Granados E., Misbah S., and Chapel H. 2010. Infection outcomes in patients with common variable immunodeficiency disorders: Relationship to immunoglobulin therapy over 22 years. J. Allergy Clin. Immunol. 125:1354–1360.e4.
Picard C., Al-Herz W., Bousfiha A., Casanova J.L., Chatila T., Conley M.E., Cunningham-Rundles C., Etzioni A., Holland S.M., Klein C., Nonoyama S., Ochs H.D., Oksenhendler E., Puck J.M., Sullivan K.E., Tang M.L., Franco J.L., and Gaspar H.B. 2015. Primary immunodeficiency diseases: An update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J. Clin. Immunol. 35:696–726.
Roifman C.M., Levison H., and Gelfand E.W. 1987. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet. 329:1075–1077.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 1 • March 2019
Pages: 31 - 33
History
Received: 31 January 2019
Accepted: 19 February 2019
Accepted manuscript online: 25 February 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
StephenBetschel, RaeBrager, AlisonHaynes, ThomasIssekutz, Vy Hong-DiepKim, BruceMazer, ChristineMcCusker, Chaim M.Roifman, TamarRubin, GordonSussman, StuartTurvey, and SusanWaserman. 2019. Report of the National Immunoglobulin Replacement Expert Committee: algorithm for diagnosis of immunodeficiency requiring antibody replacement therapy. LymphoSign Journal.
6(1): 31-33. https://doi.org/10.14785/lymphosign-2019-0003
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. COVID-19 post-vaccination recommendations for primary immunodeficiency
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
