Chronic granulomatous disease 2018: advances in pathophysiology and clinical management
Abstract
Chronic granulomatous disease (CGD) is a rare immunodeficiency disorder of phagocytic cells resulting in failure to kill a characteristic spectrum of bacteria and fungi and to resolve inflammation. The last few years have witnessed major advances in pathogenesis and clinical management of the disease:
1.
Better understanding of 3 physiologic anti-inflammatory functions of NADPH oxidase-derived reactive oxygen species: Promotion of the clearance of dying host cells, suppression of inflammasomes, and regulation of type I interferon signalling. This insight is opening new avenues for targeted drug interventions.
2.
Advances in reduced intensity conditioning (RIC) for allogeneic hematopoietic stem cell transplantation (HSCT) make it a promising and safe procedure even for fragile patients with ongoing severe infection or hyperinflammation.
3.
Encouraging early data of a multicenter trial of gene-replacement therapy using a self-inactivated lentiviral vector.
Combining targeted anti-infectious/anti-inflammatory measures and considering extended indications for curative HSCT are key to improving patient outcome further. Gene therapy will likely become a viable option for disease correction, but long-term assessment is not yet possible.
Statement of novelty: We discuss important advances in pathogenesis and treatment of CGD that will change our approach to clinical management.
Introduction to chronic granulomatous disease (CGD)
CGD is a rare inherited deficiency of the innate immune system affecting 4–5 per million live births in North America. The disease is characterized both by severe recurrent bacterial and fungal infections as well as hyperinflammatory complications (most evident in the pulmonary, gastrointestinal, and urinary tracts). CGD results from the absence or malfunction of any of the 5 NADPH oxidase subunits in phagocytic cells. Physiologically, after stimulation of neutrophils, monocytes, and macrophages, the activated oxidase transfers electrons from NADPH to molecular oxygen generating short-lived superoxide (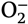 ), the so-called oxidative burst, which is converted into microbicidal reactive oxygen species (ROS, e.g., highly diffusible hydrogen peroxide; H2O2) (Figure 1). In CGD, generation of and formation of H2O2 are defective. The quantitative dihydrorhodamine 123 (DHR) flow cytometry assay (detecting H2O2) is today’s most accurate diagnostic test for CGD (Roos et al. 2017), while the qualitative (microscopical) and less discriminant nitroblue tetrazolium (NBT) dye test (detecting ) is still in clinical use.
), the so-called oxidative burst, which is converted into microbicidal reactive oxygen species (ROS, e.g., highly diffusible hydrogen peroxide; H2O2) (Figure 1). In CGD, generation of and formation of H2O2 are defective. The quantitative dihydrorhodamine 123 (DHR) flow cytometry assay (detecting H2O2) is today’s most accurate diagnostic test for CGD (Roos et al. 2017), while the qualitative (microscopical) and less discriminant nitroblue tetrazolium (NBT) dye test (detecting ) is still in clinical use.
Figure 1:
Etiology and pathophysioloy of the disease
New knowledge of the multiple physiologic functions of the phagocyte NADPH oxidase (phox) complex can now define CGD as a dual genetic defect of pathogen clearance by neutrophils and of clearance of dying host cells by macrophages. This insight is important for understanding the seemingly paradoxical association between primary immunodeficiency and autoinflammation and is opening new opportunities for targeted drug treatments.
Genetic defects
In North America, 70% of CGD cases are caused by loss-of-function mutations in the X-linked gene encoding gp91phox, the electron transferase subunit of the NADPH complex (Figure 1). 30% of cases are caused by biallelic mutations in either of 3 autosomal recessive (A/R) genes encoding cytosolic proteins (p47, p67, p40phox) which regulate gp91phox after their translocation to the plasma membrane, or in the gene coding for the membrane-bound p22phox protein that stabilizes gp91phox. A/R mutations in the gene encoding p47phox are the second most common cause of CGD, observed in 20% of cases in North America.
In regions with high rates of consanguinity (e.g., North Africa, the Arab world, Israel, and Turkey) A/R forms of CGD predominate over the X-linked recessive (XR) form. An additional factor contributing to the A/R CGD predominance are founder effects in specific populations. Altogether the mutations identified in A/R CGD patients in these regions are mostly homozygous, indicating that both parents contributed an identical, mutated allele that causes the disease (Wolach et al. 2017).
To the above list of 5 genetic NADPH oxidase defects, a novel sixth cause of CGD has to be added, namely A/R loss-of-function mutations in the CYBC1 gene coding for EROS. This hitherto undescribed transmembrane protein is highly expressed in neutrophils, monocytes, and macrophages. It plays a central role as chaperone, critical for expression of the gp91phox-p22phox heterodimer in cell and phagosomal membranes, and is thereby essential for ROS generation. EROS-deficient mice quickly succumb to infection (Thomas et al. 2017), and in a consanguineous family an EROS-deficient A/R CGD patient was found with both infectious and autoinflammatory manifestations, who subsequently underwent successful hematopoietic stem cell transplantation (HSCT) (Thomas et al. 2019). Eight additional homozygous CYBC1 deficient individuals with signs of CGD were identified in the Icelandic population likely reflecting a founder effect (Arnadottir et al. 2018).
The disease-causing gene mutation should be determined in every CGD patient. This is essential for genetic counseling and important for prognostication in CGD (Kuhns et al. 2010): Residual  generation (as in most p47phox and p40phox deficiencies (van de Geer et al. 2018)) presents a lower risk for infections and mortality compared to absent formation (as in most cases of gp91phox deficiency).
generation (as in most p47phox and p40phox deficiencies (van de Geer et al. 2018)) presents a lower risk for infections and mortality compared to absent formation (as in most cases of gp91phox deficiency).
In gp91phox deficiency, a deletion extending into the telomeric XK gene should be excluded when patients manifest red blood cell acanthocytosis and absent expression of the Kx blood group antigen. In patients with this McLeod neuroacanthosis syndrome, transfusions of ubiquitous Kell antigen positive blood will cause strong reactions due to generation of anti-Kx antibodies (Jung et al. 2007).
Impaired microbial killing
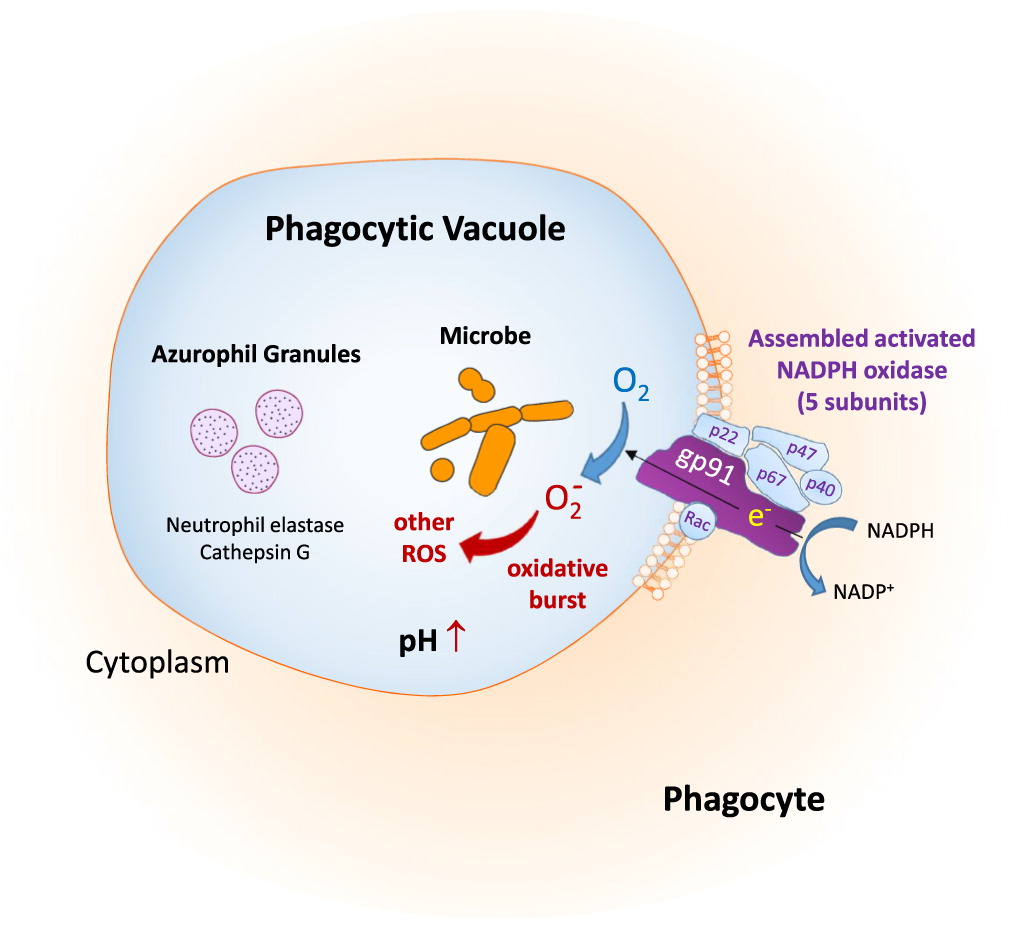
In healthy individuals, the NADPH oxidase in neutrophils, apart from being directly responsible for production of microbicidal ROS, is indirectly responsible for liberation and activation of complementary microbicidal proteases (e.g., elastase and cathepsin G) from primary (azurophilic) granules (Figure 1). Both mechanisms collaborate in killing/digestion of pathogens entrapped by neutrophils in phagocytic vacuoles, a process deficient in CGD patients (Reeves et al. 2002).
A fraction of neutrophils producing finally disintegrate their own intracellular membranes and release decondensed chromatin (DNA/histones) together with microbicidal granule proteins into the extracellular space, forming web-like neutrophil extracellular traps (NETs) (Figure 2) (Sollberger et al. 2018). We now know that pus consists mostly of neutrophils surrounded by NETs. Released NETs continue the killing process for hours and can also trap and kill microbes that are too big to be phagocytosed (e.g., bacterial aggregates and fungal hyphae). This ancient antimicrobial defense is deficient in CGD patients (Fuchs et al. 2007) and can be reversed by gene therapy (Bianchi et al. 2011).
Figure 2:

Excessive inflammation
In healthy individuals, dying (apoptotic) cells are rapidly cleared by macrophages in an “immunologically silent” way therefore preventing hyperinflammation by the debris. Apoptotic neutrophils externalize oxidized phosphatidyl serine (oxPS) on their surface membrane which is then recognized through oxPS receptors on macrophages, triggering neutrophil uptake into phagocytic vacuoles (Figure 3) (Matsura 2014).
Figure 3:
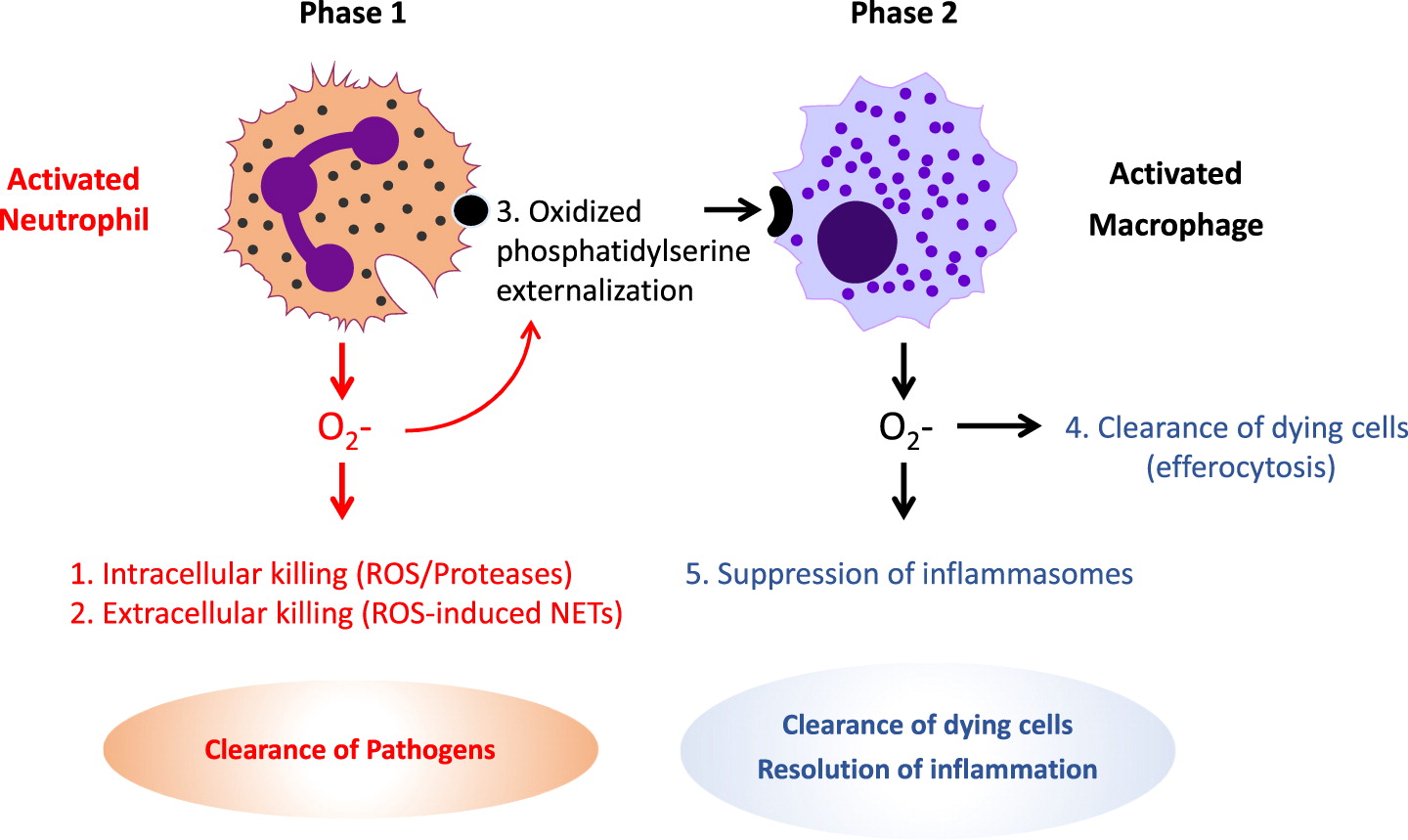
The subsequent clearance of apoptotic neutrophils within macrophages, also called efferocytosis (Latin effere = “to carry to the grave”), ultimately resolves inflammation. Following uptake, LC3-proteins of the autophagy system are recruited to phagosomes in an NADPH-oxidease dependent manner, thereby enhancing their fusion with lysosomes and facilitating controlled degradation of the cargo (Figure 4) (Heckmann et al. 2017). LC3-associated phagocytosis is accompanied by release of anti-inflammatory cytokines (e.g., interleukin (IL)-10).
Figure 4:
In CGD patients infectious foci stimulate granuloma formation. Chronic granulomatous inflammation may compromise vital organs and account for additional morbidity. Apoptosis of neutrophils (Sanford et al. 2006) and their clearance by macrophages are delayed in CGD (Bagaitkar et al. 2018). Defective clearance of dying cells leads to excess generation of pro-inflammatory cytokines (e.g., IL-1 beta and TNF alpha) and autoinflammation (Figure 5). In CGD patients, defective efferocytosis of human monocytes can be restored during experimental short term treatment with pioglitazone (PIO), a licensed anti-diabetic drug mediating resolution of inflammation. This treatment is accompanied by enhanced phagocyte mitochondrial ROS production, which bypasses the NADPH oxidase defect (Fernandez-Boyanapalli et al. 2015a). Since PIO also restores bactericidal capacity in murine CGD (Fernandez-Boyanapalli et al. 2015b), it was used in an infant with CGD to overcome refractory bacterial lung abscesses before succesful HSCT (Migliavacca et al. 2016). Efficacy and safety of this intervention require further exploration of PIO in therapy-refractory CGD patients with an indication for HSCT.
Figure 5:
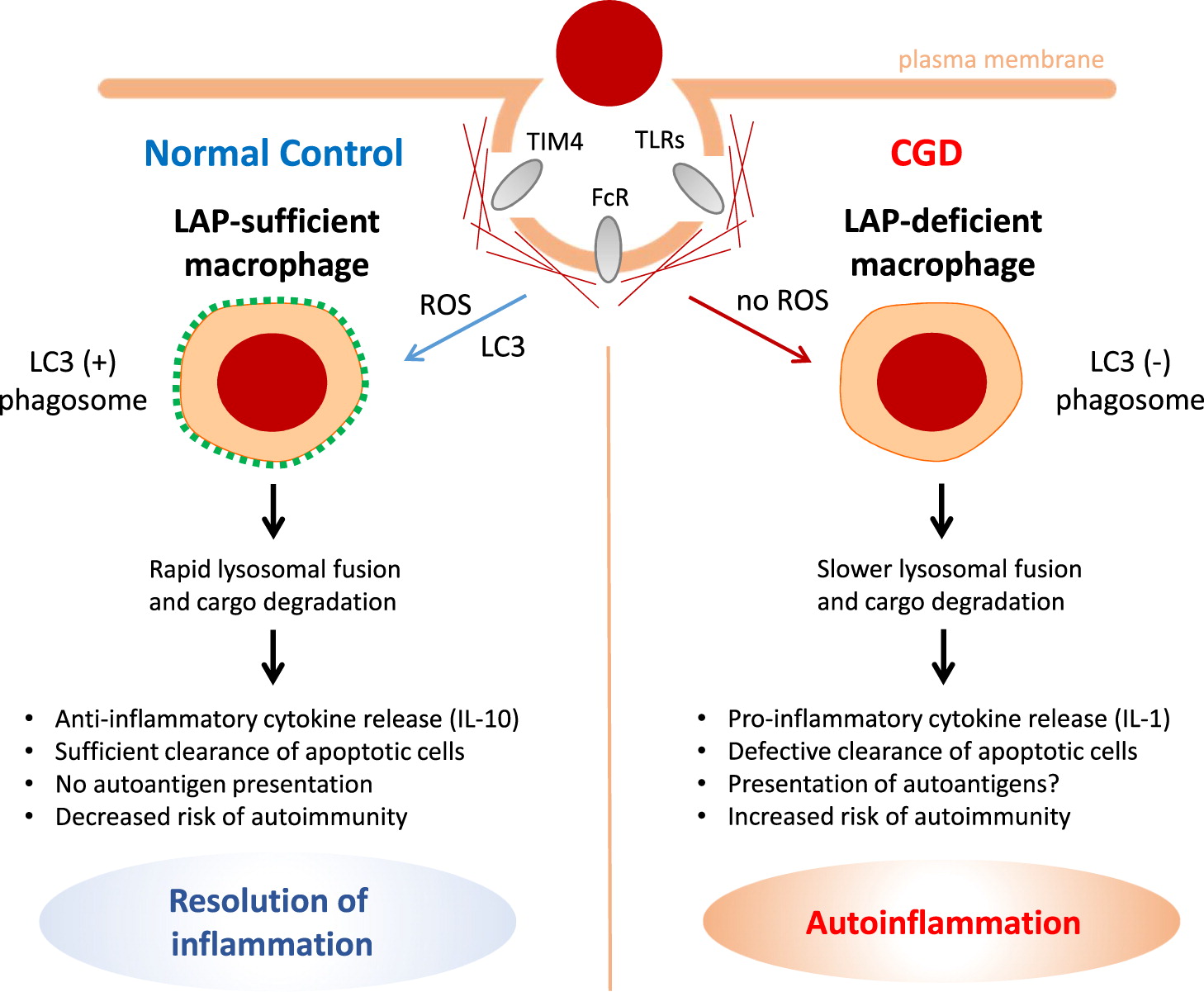
Clinical manifestations of CGD
CGD is characterized both by severe infections and hyperinflammatory manifestations. During the course of CGD the first infection occurs much earlier than the first inflammatory manifestation (at median ages of 0.9 versus 11.3 years) (Dunogué et al. 2017). While infection remains a concern in adults as the primary cause of death, inflammatory events emerge and predominate during adolescence and adulthood.
Bacterial and fungal infections
In North America (Winkelstein et al. 2000) and Europe (van den Berg et al. 2009) typical infections in CGD arise from a limited number of 5 organisms: Staphylococcus aureus (lymphadenitis, liver abscess, pneumonia rarely), Burkholderia cepacia (necrotizing pneumonia, sepsis), Serratia marcescens (sepsis, osteomyelitis), Nocardia, and Aspergillus spp. (subacute pneumonia, dissemination to brain and bone).
Invasive filamentous fungal infections are acquired through inhalation of spores, resulting in pneumonia that can spread to ribs, spine, and brain. They remain the most frequent cause of mortality in CGD. Aspergillus fumigatus is the most frequent species isolated, while Aspergillus nidulans causes more inflammatory and refractory disease (Henriet et al. 2012). Mortality is reduced by treating with azoles (voriconazole or posaconazole). Resection of infected tissue may be required in case of extension to chest wall or vertebrae.
Several emerging pathogens endemic in other regions of the world have to be added to this list and may infect also travelling CGD patients from Western countries:
•
Tuberculosis (TB) constitutes a high risk for children with CGD andresults in a complicated clinical course (Lee et al. 2008). Routine Bacillus Calmette-Guérin (BCG) vaccination in CGD causes severe disease, e.g., draining skin lesions at the vacination site, regional lymphadenitis (“BCGitis”), and more rarely disseminated disease (“BCG sepsis”) (Conti et al. 2016).
•
Non-typhoidal Salmonella is a leading cause of blood-stream infection in CGD patients in endemic regions world-wide (Lee and Lau 2017).
•
Chromobacterium violaceum, a gram negative bacillus, confined to (sub)tropical climates and encountered in brackish waters, causes sepsis in exposed CGD patients (Sirinavin et al. 2005).
•
Burkholderia pseudomallei, a water/soil bacterium found in (sub)tropical regions causes systemic melioidosis. It has been identified in CGD children of South East Asia. They acquire the infection during work in rice-fields via skin wounds, inhalation, drowning, or after heavy monsoon rains (Lee and Lau 2017).
•
Leishmania infantum, an intracellular protozoon endemic on the Mediterranian coast, has caused visceral leishmaniasis-associated hemophagocytic syndrome in Portuguese and Spanish CGD patients (Martín et al. 2009).
Inflammatory manifestations
The classic inflammatory complications of CGD are most prominent in the pulmonary, gastrointestinal, and urinary tracts (e.g., as interstitial lung disease, granulomatous colitis, and granulomatous cystitis). Granulomatous colitis in CGD mimicks Crohn’s disease and affects up to one half of CGD patients (Marciano et al. 2004). Colonoscopy permits diagnostic biopsies revealing epitheloid granulomas and pigment-laden macrophages. Empiric initial therapy is based on corticosteroids.
To this list, 2 emerging complications have to be added:
•
Fulminant mulch pneumonitis, an emergency caused by massive inhalation of aerosols from mulch, compost, or dead leaves containing Aspergillus spores. The excessive inflammatory response to the spores results in miliary infiltrates and hypoxia requiring ventilation. An intravenous combination of voriconazole and methylprednisolone is life-saving (Siddiqui et al. 2007).
•
Non-cirrhotic portal hypertension following liver abscesses: obliteration of central/portal veins and nodular regenerative hyperplasia can ultimately lead to portal hypertension (Hussain et al. 2007). Liver abscesses, mostly by S. aureus, in the context of prolonged fever should be treated by antibiotics in combination with steroids to avoid excess surgical intervention (Straughan et al. 2018).
Surgical sites in CGD often become infected and heal very slowly with fistulas. Sutures should not be removed early and drains be left for a prolonged period. Excessive wound granulation with dehiscence responds to steroids.
In addition to autoinflammation, there is an increased risk of autoimmune disease in CGD with a focus on syndromes that resemble lupus. In the national US registry of more than 350 CGD patients, 0.5% of patients met diagnostic criteria for SLE and 2.7% criteria for discoid lupus (Winkelstein et al. 2000).
Classic SLE is characterised by overexpression of type I interferon pathway transcripts that are also highly expressed in CGD patients and in p47phox deficient mice (Holmdahl et al. 2016; Thomas 2017). This finding would implicate NADPH oxidase-derived ROS as a feedback mechanism dampening activation of the type I interferon pathway and possibly preventing autoimmunity.
Clinical presentation in adults
In the first nation-wide retrospective study focusing on the long-term outcome of a French pediatric CGD cohort of 80 patients, the grown-up CGD patients displayed similar rates and characteristics of severe infections and inflammatory episodes as in childhood (Dunogué et al. 2017). Main sequelae of pediatric CGD observed in adulthood were:
1.
Growth failure as a consequence of serious infections, inflammation, and of repeated steroid treatments.
2.
Chronic dyspnea from restrictive respiratory failure after repeated lung infections.
3.
Chronic digestive complications from episodes of inflammatory colitis, bowel stenosis, and perianal fistulas with substantial impact on quality of life.
The social consequences of the above complications and repeated hospital admissions were serious with poor educational achievement in half of the patients. Proper surveillance and follow up are required for adult CGD patients, especially during the crucial transition from pediatric to adult medical departments. Regular follow up should be performed to detect and properly manage occult infections, effectively treat inflammatory events, and prevent long-term complications (Thomsen et al. 2016).
Clinical presentation in carriers of X-linked CGD
While many female carriers of X-linked CGD are asymptomatic, some become symptomatic with severe or recurrent infections, or autoimmune (e.g., discoid lupus erythematosus) or inflammatory (e.g., inflammatory bowel disease) manifestations. In female carriers of XR-CGD the CYBB gene, encoding gp91phox, is silenced randomly in each cell early in development allowing expression of only 1 X-chromosome (so-called lyonization). Neutrophils with inactivation of the CYBB-mutated X-chromosome will have normal production whereas cells with inactivation of the normal X-chromosome will have a CGD phenotype.
In a large study of 93 females with XR-CGD carrier state (Marciano et al. 2018), carriers with CGD type infections showed a strong correlation between risk of infections and skewed inactivation of the wild type allele (with <20% forming cells). From these data it seems prudent to consider trimethoprim/sulfamethoxazole prophylaxis in carriers when the %DHR value is <20%. In contrast, susceptibility to autoimmune/inflammatory manifestations in the XR-CGD carrier state was unrelated to the amount of generation. The association of autoimmune phenomena with the carrier state per se, nevertheless, suggests that XR-CGD carriers in general are not ideal donors for HSCT.
Conventional treatment of CGD
Clinical management of CGD has been reviewed before (Leiding and Holland 2016; Seger et al. 2017).
General health care
Common sense measures are sometimes neglected and have to be re-emphasized to patients and parents:
- Wounds should be washed well and rinsed with antiseptic solutions (e.g., 2% H2O2 or Betadine).
- Extensive dental work/surgery associated with bacteremia should be covered with additional antibiotics, e.g., amoxicillin/clavulanic acid.
- Pulmonary infections can be prevented by refraining from smoking, not using bedside humidifiers and avoiding sources of Aspergillus spores (animal stables, garden work, and constructions sites).
- Risk of perirectal abscesses can be diminished by avoiding rectal manipulations, e.g., suppositories or taking rectal temperature.
- All routine immunizations should be given to CGD patients except live bacterial vaccines (BCG and Salmonella typhi vaccines, contraindicated in CGD). Measles and varicella live vaccines as well as yearly influenza vaccine are safe and indicated to prevent potentially lethal bacterial superinfections.
- CGD patients traveling to or residing in endemic regions should know about potential infections and adopt preventive measures as advised by specialists in travel medicine (Lee and Lau 2017).
Antimicrobial prophylaxis
The cornerstone of clinical care is mainly based on retrospective studies and consists of lifelong antibacterial and antifungal prophylaxis with intracellularly active microbicidal agents. Lipophilic co-trimoxazole (trimethoprim/sulfamethoxazole, TMP/SMX) results in a marked reduction of serious bacterial infections and abscess drainages (Margolis et al. 1990). It is recommended at 5 mg/kg/day, TMP up to 160 mg daily.
For antifungal prophylaxis the lipophilic itraconazole is the drug of choice with high activity against Aspergillus spp. (Gallin et al. 2003). Itraconazole is recommended up to a maximum of 200 mg once daily. For optimal bioavailability, itraconazole capsules should be taken at 5 mg/kg/day with food, while itraconazole solution should be taken at 2.5 mg/kg/day in fasting condition.
In addition to the above antimicrobials, interferon gamma (IFNγ) is part of the routine prophylaxis regimen in US centers, while most European experts use IFNγ only in selected cases. The dose is 50 ug/m2 s.c. 3×/week. This prophylactic regimen is based on a prospective multicenter randomized placebo-controlled trial of IFNγ in 128 CGD patients performed before the advent of antifungal prophylaxis with itraconazole. The trial resulted in reduction of the frequency of mainly severe bacterial infections >70% (The International Chronic Granulomatous Disease Cooperative Study Group 1991). There were no improvements in NADPH oxidase function nor a significant efficacy in preventing Aspergillus infections during the limited study period. A later prospective non-randomized long term (lasting 5 years) Italian multicenter study of 35 CGD patients, comparing treatment with TMP/SMX and itraconazole alone versus addition of IFNγ, showed no difference in the rates of severe infection (Martire et al. 2008). The exact mechanism of how IFNγ exerts its effect in CGD is still unknown, adding to the debate over its utility. Since IFNγ upregulates human leukocyte antigen (HLA) expression, IFNγ prophylaxis has to be stopped at least 4 weeks before HSCT.
Treatment of serious infections
Significant rises in C-reactive protein (CRP) should prompt evaluation for infection, including appropriate imaging and pathogen identification. In the case of fever and (or) persistent cough, a liver abscess, Salmonella and Burkholderia septicemia as well as Aspergillus pneumonia (inhalational miliary or focal invasive) have to be excluded first. CT or MRI imaging should be followed until resolution of infections. A definitive microbiological diagnosis by tissue biopsies is essential for proper treatment. An appropriate sample may require fine needle aspiration or percutaneous drainage of liquid pus.
Recommended empiric initial therapy is based on limited clinical data because of the rarity of CGD. Consultation with an experienced infectious disease specialist is strongly advised. Antibiotics chosen for initial therapy should cover a broad spectrum of gram negative bacteria (including Burkholderia spp. and Serratia marcescens) as well as gram-positive organisms (including S. aureus and Nocardia spp.). Useful first-line agents with an appropriate antimicrobial spectrum are meropenem PLUS vancomycin added in regions with methicillin-resistant S. aureus. In case of failure to respond within 24–48 h, an antimycotic drug (e.g., voriconazole) may be needed. Initial therapy is tailored once culture/susceptibility and histopathology are known. As infections often respond slowly, intravenous treatment must be followed by prolonged oral treatment, sometimes continued over months.
Consider addition of steroids in case of fulminant Aspergillosis or severe Nocardia infection not responsive to appropriate antibiotic therapy (Siddiqui et al. 2007; Freeman et al. 2011).
Treatment of inflammatory complications
Antimicrobial prophylaxis decreases the risk of severe infections, but not the risk of inflammatory manifestations which can affect >50% of patients (especially those with XR-CGD). Management of inflammatory complications is challenging, as treatment with antiinflammatory/immunosuppressive agents increase the risk of infection. Treatment of inflammatory manifestations in CGD has been reviewed before (Magnani and Mahlaoui 2016).
Short courses of corticosteroids followed by gradual tapering are required in acute granulomatous exacerbations of the bowel (e.g., gastric outlet obstruction), the urinary tract (e.g., ureteral obstruction) and the lung (inhalative acute miliary pneumonia). Long-term treatment of granulomatous colitis follows treatment options for Crohn’s disease under the cover of the routine antimicrobial prophylaxis in CGD. Recommended first-line therapy in severe CGD-colitis cases is prednisone (1 mg/kg/day) for 1–2 weeks with slow tapering over 1–2 months to 0.1–0.25 mg/kg/day (Leiding and Holland 2016). Steroids inhibit granuloma formation by suppressing proinflammatory cytokine production and TNFα-dependent fusion of macrophages into multinucleated giant cells (Maltesen et al. 2010). Steroid dosage during taper can be adjusted to the severity of colonic inflammation by following fecal levels of calprotectin released from apoptotic neutrophils (Nakazawa et al. 2017).
Steroids are effective in severe CGD-colitis, but when taken off treatment relapse is high. Some patients become steroid-dependent requiring second line therapies (including immunosuppressants and biologic agents). Patients with refractory colitis necessitate a multi-step approach and may become eligible for allogeneic HSCT.
Azathioprine, an immunosuppressant, is used as second line therapy to maintain remission in steroid-dependent cases and to improve outcomes in patients with fistulating disease.
Thalidomide blocks nuclear localization of NFκB, thus inhibiting the production of inflammatory cytokines. It has been used by the Paris group for treatment of refractory colitis with complete clinical responses in 4/6 patients after 6 months (Noel et al. 2013) and was not associated with increased risk of infections.
Infliximab, a monoclonal antibody to TNFα, was used by the NIH group in 5 CGD patients with steroid-refractory colitis. It is effective, but resulted in severe intercurrent infections with CGD pathogens in all 5 and death in 2, precluding long-term therapy in CGD (Uzel et al. 2010).
Use of Anakinra, an IL-1-receptor antagonist, has also been reported in CGD patients with refractory colitis. Its efficacy has been contested by the NIH group, when treatment of 5 CGD patients with severe colitis led only to marginal or no benefit (Hahn et al. 2015).
Allogeneic HSCT is the only curative treatment for refractory colitis in CGD. HSCT can save patients from extensive colon surgery, risky because of poor wound healing, anastomosis complications and fistula formation. Inflammatory manifestations rapidly regress post-HSCT permitting withdrawel of steroids with a subsequent growth spurt into predetermined percentile channels in children (Seger 2010).
Curative treatments
The above prophylactic and routine therapeutic approaches are mostly supportive and depend on lifelong patient compliance. CGD thus remains a lethal disease, nowadays at an adult age. The ultimate goal is to develop safe curative approaches, e.g., allogeneic HSCT regimens with reduced-intensity conditioning (RIC) and autologous stem cell gene therapies (GT) for CGD patients without a suitable HLA-compatible donor.
Allogeneic HSCT
CGD is a proinflammatory disease with a high risk of alloreactivity (rejection and GvHD) after myeloablative cytotoxic marrow conditioning. Therefore, antithymocyte globulin in HLA-matched sibling transplantation and alemtuzumab in matched unrelated (MUD) transplantation were introduced for balanced in vivo depletion of alloreactive recipient and donor T cells. In addition, toxic myeloablative conditioning was replaced by a sub-myeloablative RIC regimen to decrease tissue damage and reduce release of proinflammatory cytokines. These 2 measures allowed safe transplantation even in high-risk CGD patients with ongoing infection/inflammation and in younger adults. Pre-existing infections and chronic inflammatory lesions cleared in all engrafted survivors. Even children with severe lung restriction improved their lung function slowly, normalizing decreased oxygen saturation, reversing clubbed fingers and toes, and manifesting a growth spurt (reviewed in Seger 2010).
To further optimize the RIC protocol, targeted drug monitoring (TDM) was introduced. Low-dose busulfan is administered, serum busulfan levels are measured, and the cumulative area-under-the-concentration curve was individually adjusted real-time to a sub-myeloablative target range of 45–65 mg/Lxh. An international prospective CGD/HSCT trial using this Zürich RIC protocol was performed at 16 centers in 10 countries (Güngör et al. 2014). 56 CGD patients aged 0–40 years were enrolled of whom 42 patients had high-risk features and 25 were adolescents or young adults. 2-year probability of overall survival (OS) was 96% and event-free survival (EFS) was 91%. Equivalent outcomes between matched siblings and MUDs were observed. Incidences of acute graft versus host disease (GvHD) and limited chronic GvHD (cGvHD), as well as an incidence of 5% of graft failure were low. Excellent myeloid donor chimerism (>90%) was documented in 93% of surviving patients.
Because of its efficacy and favorable toxicity the low busulfan RIC regimen is a promising treatment modality for CGD, however, requires good lab facilities for real-time TDM. In the absence of TDM facilities, the Zürich RIC protocol may still be used by aiming at a higher submyeloablative target of 65 mg/Lxh with the help of a recent busulfan dosing nomogram (Bartelink et al. 2012).
Another reduced toxicity conditioning (RTC) regimen based on myeloablative treosulfan (available in Europe) was used by the Newcastle group and has also achieved good survival rates in a large retrospective CGD study (Morillo-Gutierrez et al. 2016). Secondary graft failure in 12% of the 70 patients requires further investigation, especially with regard to the durability of myeloid chimerism.
The decision for or against allogeneic HSCT should be made early in childhood. Patients with gp91phox mutations and absent production have a survival of only 50% beyond 40 years of age compared to patients with p47phox mutations and residual H2O2 generation (over 80% survival beyond 40 years) (Kuhns et al. 2010). Our algorithm for patient selection and HSCT timing in CGD (Figure 6) is based on 4 parameters, early HLA-typing, quantitative production, mutation analysis, and the individual clinical course. Provided a suitable HLA-compatible MSD/MUD donor has been found, 3 situations have to be analysed:
1.
If there is completely absent 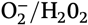 production early HSCT can be recommended for any type of CGD (XR-CGD or A/R CGD)(because of the high long-term risk of therapy-refractory infection/inflammation and death in any CGD patient with no oxidase function).
production early HSCT can be recommended for any type of CGD (XR-CGD or A/R CGD)(because of the high long-term risk of therapy-refractory infection/inflammation and death in any CGD patient with no oxidase function).
2.
In case of residual generation with better long-term outcome watchful waiting on medical therapy may be recommended for any type of CGD while observing the individual clinical course for a clinical HSCT indication.
3.
In case of late presentation for HSCT after an eventful therapy-refractory clinical course with important organ sequelae, salvage HSCT can still be successful but needs to be discussed carefully by a board of experienced transplanters.
Figure 6:
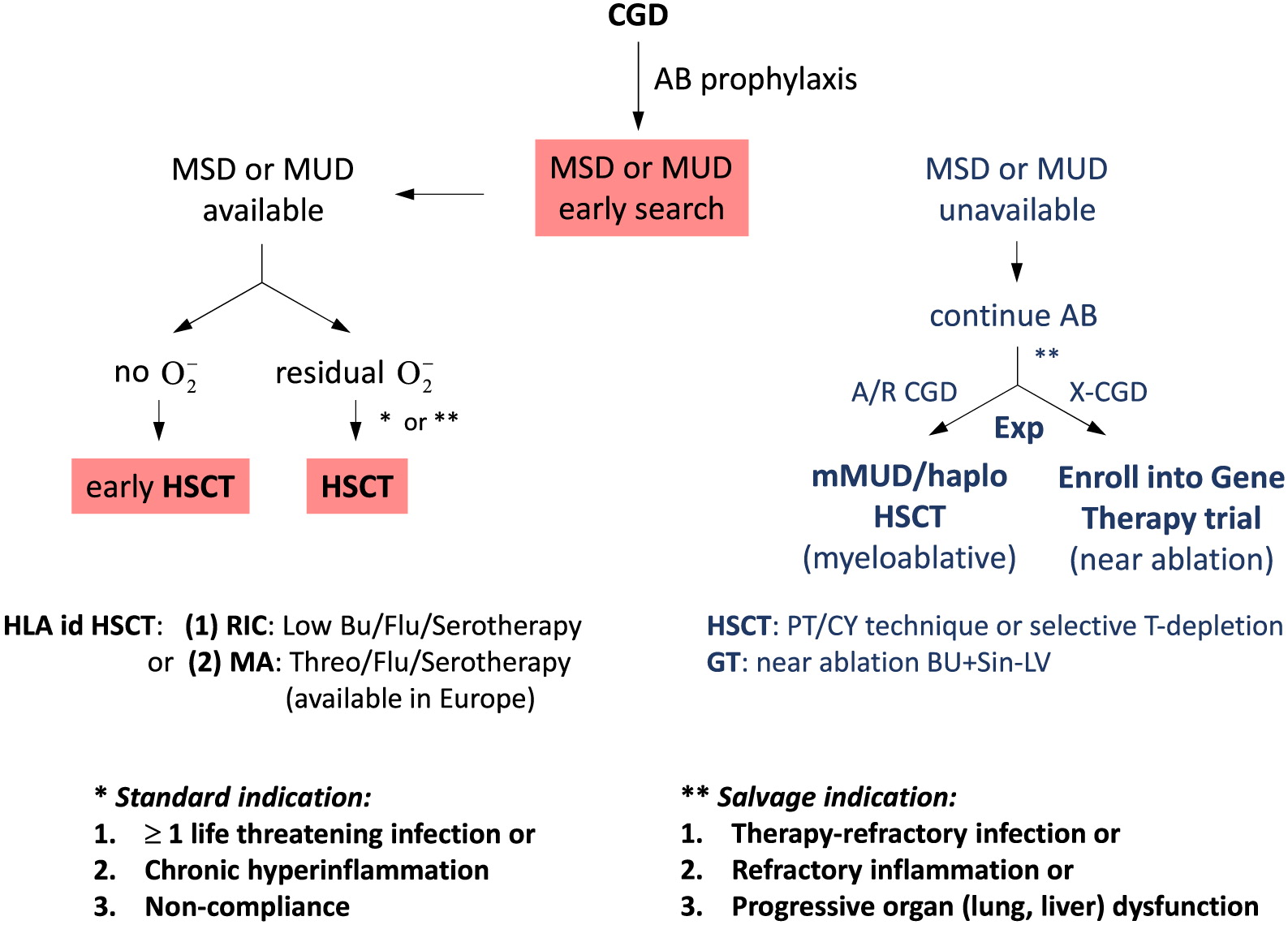
Two recent reports from Sweden (Åhlin et al. 2013) and UK (Cole et al. 2013) comparing HSCT versus conventional treatment support HSCT as being the preferable treatment for severe CGD. Children not undergoing transplantation have more serious infections, surgical interventions, hospital admissions and lower height for age compared with post-HSCT children. Long-term post-HSCT survival and quality of life data in adult CGD are not yet available.
Therapies under investigation
HLA-haploidentical HSCT
Transplants with haploidentical donors present greater risk of graft rejection and GvHD. The current use of CD34+ selected stem cell grafts (with 4–5 log donor T cell depletion from the transplant to avoid severe GvHD) results in 4–6 months of profound T cell deficiency. This non-selective, global T cell depletion technique is accompanied by risks of graft failure/rejection as well as severe viral reactivation until thymopoiesis is established. Three novel approaches to “graft engineering” have been developed in order to minimize these 2 risks and are now improving outcomes for PID patients with haploidentical parent donor grafts.
1.
A new protocol of selective depletion of alloreactive T-cells in vivo, on days +3/+4 post-HSCT by high-dose cyclophosphamide (at 50 mg/kg/day) presents a promising alternative (Luznik et al. 2010). Cyclophosphamide (Cy) is non-toxic to stem cells and kills rapidly deviding alloreactive T cells post transplantation (PT), while sparing resting T cells with specificity for infectious agents. PT/Cy controls GvHD without affecting engraftment and allows more rapid reconstitution of donor immunity to opportunistic infections. This promising new technique has been successfully applied in a few CGD patients (Parta et al. 2015; Regueiro-García et al. 2018) and should now be investigated in a prospective multicenter study. The easy logistics of PT/Cy are an asset in countries with no access to an unrelated donor registry and lacking a sophisticated laboratory for graft manipulations required for conventional T cell depletion of haploidentical grafts.
2.
In countries with available laboratory technology another refined in vitro method of selective T cell depletion is increasingly used for removal of GvHD-inducing CD3+TCR alpha/beta+ T cells from HLA-haploidentical grafts. This new depletion technique retains TCR gamma/delta+ T cells which can promote engraftment without causing GvHD and might alleviate viral reactivation. In 3 landmark PID studies using CD3+TCR alpha/beta+/CD19+ depletion a combined overall transplantation survival of 90% was seen, including 4 successfully transplanted CGD patients (e.g., Shah et al. 2018). However, a significant rate of viral infections was still observed, despite the presence of TCR gamma/delta+ T lymphocytes.
3.
A solution to viral disease is the adoptive transfer of virus-specific memory T cells from an antigen-experienced donor. Prior to infusion the donor T cells have to be depleted of GvHD-inducing CD45 RA+ naive T cells, while retaining their anti-infectious memory T cell repertoire. CD45 RA+ depleted donor lymphocyte infusions (DLIs) can safely be administered at low T cell doses in monthly intervals post HSCT, escalating to 100 × 103 T cells/kg in haploidentical recipients. Among 31 patients with absent CMV-specific immune reactivity at baseline significant expansion of CMV-specific T cells after 3 DLIs was demonstrated in 20 within 100 days (Maschan et al. 2018). CD45 RA-depleted memory DLIs thus constitute a simple and likely efficient measure to prevent viral reactivation in the setting of TCR alpha/beta T cell depleted grafts.
Stem cell gene therapy
CGD remains an attractive though difficult target for autologous stem cell gene therapy (GT). Functional correction of as few as 10%–20% of neutrophils should be sufficient to prevent CGD typical infections based on the experience in X-linked CGD carriers (Marciano et al. 2018). The major obstacle however is the lack of selective growth advantage of gene-transduced stem cells. Previous protocols of unregulated gene addition mediated by retroviral vectors have either resulted in graft failure or development of a myelodysplastic syndrome due to transactivation of the MDS1/EVI1 protooncogene by gamma-retroviral insertions (Stein et al. 2010). Early data of an ongoing prospective clinical trial in XR-CGD (based on a new self-inactivating lentiviral vector with an internal myelospecific chimeric promoter driving gp91phox expression (Santilli et al. 2011)) indicate a safer integration profile with no evidence of transactivation of nearby cellular genes/clonal expansion and stable oxidase activity in >20% of neutrophils in the first treated CGD patients (Thrasher 2018).
Following the excellent outcome of recent clinical trials of viral-mediated gene therapy of HSCs for several PIDs resulting in long-term constitutive gene expression, search efforts are now directed to site-specific gene editing allowing expression of corrective DNA sequences at the native site under physiologic regulation or in a genomic “safe harbour” (Keller et al. 2018). Taking the advances in GT and HSCT together, one can be cautiously optimistic about further important advances in the next 5–10 years on the long road to optimal curative therapies for CGD patients lacking a suitable transplantation donor.
Abbreviations
- AR
- autosomal recessive
- CGD
- chronic granulomatous disease
- GT
- gene therapy
- GvHD
- Graft-versus-host disease
- HSCT
- hematopoietic stem cell transplantation
- IFNγ
- interferon gamma
- IL-1β
- interleukin 1 beta
- MA
- myeloablation
- NET
- neutrophil extracellular trap
- superoxide anion
- oxPS
- oxidized phosphatidylserine
- PHOX
- phagocyte NADPH oxidase
- PIO
- pioglitazone
- PT/CY
- post-transplant cyclophosphamide
- RIC
- reduced intensity conditioning
- ROS
- reactive oxygen species
- TDM
- targeted drug monitoring
- TMP-SMX
- trimethoprim-sulfamethoxazole
- TNFα
- tumor necrosis factor alpha
- XR
- X-linked recessive
Acknowledgements
Dedicated to Andrea Gressani and Luciano Vassalli: Affected by CGD and lacking a marrow donor, their hope for definitive therapy set spurs to our team.
REFERENCES
Åhlin A., Fugeläng J., de Boer M., Ringden O., Fasth A., and Winiarski J. 2013. Chronic granulomatous disease—Haematopoietic stem cell transplantation versus conventional treatment. Acta Paediatr. 102:1087–1094.
Arnadottir G.A., Norddahl G.L., Gudmundsdottir S., Agustsdottir A.B., Sigurdsson S., Jensson B.O., Bjarnadottir K., Theodors F., Benonisdottir S., Ivarsdottir E.V., Oddsson A., Kristjansson R.P., Sulem G., Alexandersson K.F., Juliusdottir T., Gudmundsson K.R., Saemundsdottir J., Jonasdottir A., Jonasdottir A., Sigurdsson A., Manzanillo P., Gudjonsson S.A., Thorisson G.A., Magnusson O.Th., Masson G., Orvar K.B., Holm H., Bjornsson S., Arngrimsson R., Gudbjartsson D.F., Thorsteinsdottir U., Jonsdottir I., Haraldsson A., Sulem P., and Stefansson K. 2018. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes CGD. Nat. Commun. 9:4447.
Bagaitkar J., Huang J., Zeng M.Y., Pech N.K., Monlish D.A., Perez-Zapata L.J., Miralda I., Schuettpelz L.G., and Dinauer M.C. 2018. NADPH oxidase activation regulates apoptotic neutrophil clearance by murine macrophages. Blood. 131:2367–2378.
Bartelink I.H., Boelens J.J., Bredius R.G., Egberts A.C., Wang C., Bierings M.B., Shaw P.J., Nath C.E., Hempel G., Zwaveling J., Danhof M., and Knibbe C.A. 2012. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: Towards individualized dosing. Clin. Pharmacokinet. 51:331–345.
Bianchi M., Niemiec M.J., Siler U., Urban C.F., and Reichenbach J. 2011. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 127:1243–1252.e7.
Cole T., Pearce M.S., Cant A.J., Cale C.M., Goldblatt D., and Gennery A.R. 2013. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 132:1150–1155.
Conti F., Lugo-Reyes S.O., Blancas Galicia L., He J., Aksu G., Borges de Oliveira E. Jr., Deswarte C., Hubeau M., Karaca N., de Suremain M., Guérin A., Baba L.A., Prando C., Guerrero G.G., Emiroglu M., Öz F.N., Yamazaki Nakashimada M.A., Gonzalez Serrano E., Espinosa S., Barlan I., Pérez N., Regairaz L., Guidos Morales H.E., Bezrodnik L., Di Giovanni D., Dbaibo G., Ailal F., Galicchio M., Oleastro M., Chemli J., Danielian S., Perez L., Ortega M.C., Soto Lavin S., Hertecant J., Anal O., Kechout N., Al-Idrissi E., ElGhazali G., Bondarenko A., Chernyshova L., Ciznar P., Herbigneaux R.M., Diabate A., Ndaga S., Konte B., Czarna A., Migaud M., Pedraza-Sánchez S., Zaidi M.B., Vogt G., Blanche S., Benmustapha I., Mansouri D., Abel L., Boisson-Dupuis S., Mahlaoui N., Bousfiha A.A., Picard C., Barbouche R., Al-Muhsen S., Espinosa-Rosales F.J., Kütükçüler N., Condino-Neto A., Casanova J.L., and Bustamante J. 2016. Mycobacterial disease in patients with chronic granulomatous disease: A retrospective analysis of 71 cases. J. Allergy Clin. Immunol. 138:241–248.e3.
Dunogué B., Pilmis B., Mahlaoui N., Elie C., Coignard-Biehler H., Amazzough K., Noël N., Salvator H., Catherinot E., Couderc L.J., Sokol H., Lanternier F., Fouyssac F., Bardet J., Bustamante J., Gougerot-Pocidalo M.A., Barlogis V., Masseau A., Durieu I., Lecuit M., Suarez F., Fischer A., Blanche S., Hermine O., and Lortholary O. 2017. Chronic granulomatous disease in patients reaching adulthood: A nationwide study in France. Clin. Infect. Dis. 64:767–775.
Fernandez-Boyanapalli R.F., Falcone E.L., Zerbe C.S., Marciano B.E., Frasch S.C., Henson P.M., Holland S.M., and Bratton D.L. 2015a. Impaired efferocytosis in human chronic granulomatous disease is reversed by pioglitazone treatment. J. Allergy Clin. Immunol. 136:1399–1401.e3.
Fernandez-Boyanapalli R.F., Frasch S.C., Thomas S.M., Malcolm K.C., Nicks M., Harbeck R.J., Jakubzick C.V., Nemenoff R., Henson P.M., Holland S.M., and Bratton D.L. 2015b. Pioglitazone restores phagocyte mitochondrial oxidants and bactericidal capacity in chronic granulomatous disease. J. Allergy Clin. Immunol. 135:517–527.e12.
Freeman A.F., Marciano B.E., Anderson V.L., Uzel G., Costas C., and Holland S.M. 2011. Corticosteroids in the treatment of severe nocardia pneumonia in chronic granulomatous disease. Pediatr. Infect. Dis. J. 30:806–808.
Fuchs T.A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., and Zychlinsky A. 2007. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176:231–241.
Gallin J.I., Alling D.W., Malech H.L., Wesley R., Koziol D., Marciano B., Eisenstein E.M., Turner M.L., DeCarlo E.S., Starling J.M., and Holland S.M. 2003. Itraconazole to prevent fungal infections in chronic granulomatous disease. N. Engl. J. Med. 348:2416–2422.
Güngör T., Teira P., Slatter M., Stussi G., Stepensky P., Moshous D., Vermont C., Ahmad I., Shaw P.J., Telles da Cunha J.M., Schlegel P.G., Hough R., Fasth A., Kentouche K., Gruhn B., Fernandes J.F., Lachance S., Bredius R., Resnick I.B., Belohradsky B.H., Gennery A., Fischer A., Gaspar H.B., Schanz U., Seger R., Rentsch K., Veys P., Haddad E., Albert M.H., Hassan M., and Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation. 2014. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: A prospective multicentre study. Lancet. 383:436–448.
Hahn K.J., Ho N., Yockey L., Kreuzberg S., Daub J., Rump A., Marciano B.E., Quezado M., Malech H.L., Holland S.M., Heller T., and Zerbe C.S. 2015. Treatment with anakinra, a recombinant IL-1 receptor antagonist, unlikely to induce lasting remission in patients with CGD colitis. Am. J. Gastroenterol. 110:938–939.
Heckmann B.L., Boada-Romero E., Cunha L.D., Magne J., and Green D.R. 2017. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429:3561–3576.
Henriet S.S.V., Verweij P.E., and Warris A. 2012. Aspergillus nidulans and chronic granulomatous disease: A unique host–pathogen interaction. J. Infect. Dis. 206:1128–1137.
Holmdahl R., Sareila O., Olsson L.M., Bäckdahl L., and Wing K. 2016. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol. Rev. 269:228–247.
Hussain N., Feld J.J., Kleiner D.E., Hoofnagle J.H., Garcia-Eulate R., Ahlawat S., Koziel D.E., Anderson V., Hilligoss D., Choyke P., Gallin J.I., Liang T.J., Malech H.L., Holland S.M., and Heller T. 2007. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology. 45:675–683.
Jung H.H., Danek A., and Frey B.M. 2007. McLeod syndrome: A neurohaematological disorder. Vox Sang. 93:112–121.
Keller M.D., Notarangelo L.D., and Malech H.L. 2018. Future of care for patients with chronic granulomatous disease: Gene therapy and targeted molecular medicine. J. Pediatric Infect. Dis. Soc. 7(Suppl 1):S40–S44.
Kuhns D.B., Alvord W.G., Heller T., Feld J.J., Pike K.M., Marciano B.E., Uzel G., DeRavin S.S., Priel D.A., Soule B.P., Zarember K.A., Malech H.L., Holland S.M., and Gallin J.I. 2010. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 363:2600–2610.
Lee, P.P., and Lau, Y.L. 2017. Endemic infections in chronic granulomatous disease. In Chronic granulomatous disease: Genetics, biology and clinical management. Edited by R.A. Seger, D. Roos, B.H. Segal, and T.W. Kuijpers. New York, NY, USA: Nova Biomedical Press. pp. 125–162.
Lee P.P., Chan K.W., Jiang L., Chen T., Li C., Lee T.L., Mak P.H., Fok S.F., Yang X., and Lau Y.L. 2008. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: A review of 17 patients living in a region endemic for tuberculosis. Pediatr. Infect. Dis. J. 27:224–230.
Leiding, J.W., and Holland, S.M. 2016. Chronic granulomatous disease. In GeneReviews® [Internet]. Edited by M.P. Adam, H.H. Ardinger, R.A. Pagon, S.E. Wallace, L.J.H. Bean, K. Stephens, and A. Amemiya. Seattle, WA, USA: University of Washington.
Luznik L., Jones R.J., and Fuchs E.J. 2010. High-dose cyclophosphamide for graft-versus-host disease prevention. Curr. Opin. Hematol. 17:493–499.
Magnani A. and Mahlaoui N. 2016. Managing inflammatory manifestations in patients with chronic granulomatous disease. Pediatr. Drugs. 18:335–345.
Maltesen H.R., Nielsen C.H., Dalbøge C.S., and Baslund B. 2010. Methylprednisolone prevents tumour necrosis factor-α-dependent multinucleated giant cell formation. Rheumatology. 49:2037–2042.
Marciano B.E., Rosenzweig S.D., Kleiner D.E., Anderson V.L., Darnell D.N., Anaya-O’Brien S., Hilligoss D.M., Malech H.L., Gallin J.I., and Holland S.M. 2004. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 114:462–468.
Marciano B.E., Zerbe C.S., Falcone E.L., Ding L., DeRavin S.S., Daub J., Kreuzburg S., Yockey L., Hunsberger S., Foruraghi L., Barnhart L.A., Matharu K., Anderson V., Darnell D.N., Frein C., Fink D.L., Lau K.P., Long Priel D.A., Gallin J.I., Malech H.L., Uzel G., Freeman A.F., Kuhns D.B., Rosenzweig S.D., and Holland S.M. 2018. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 141:365–371.
Margolis D.M., Melnick D.A., Alling D.W., and Gallin J.I. 1990. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J. Infect. Dis. 162:723–726.
Martín A., Marques L., Soler-Palacín P., Caragol I., Hernandez M., Figueras C., and Español T. 2009. Visceral leishmaniasis associated hemophagocytic syndrome in patients with chronic granulomatous disease. Pediatr. Infect. Dis. 28:753–754.
Martire B., Rondelli R., Soresina A., Pignata C., Broccoletti T., Finocchi A., Rossi P., Gattorno M., Rabusin M., Azzari C., Dellepiane R.M., Pietrogrande M.C., Trizzino A., Di Bartolomeo P., Martino S., Carpino L., Cossu F., Locatelli F., Maccario R., Pierani P., Putti M.C., Stabile A., Notarangelo L.D., Ugazio A.G., Plebani A., De Mattia D., and IPINET. 2008. Clinical features, long-term follow-up and outcome of a large cohort of patients with chronic granulomatous disease: An Italian multicenter study. Clin. Immunol. 126:155–164.
Maschan M., Blagov S, Shelikhova L., Shekhovtsova Z., Balashov D., Starichkova J., Kurnikova E., Boyakova E., Muzalevskii Y., Kazachenok A., Trakhtman P., Osipova E., Khripkova N., Zhogov V., Novichkova G., and Maschan A. 2018. Low-dose donor memory T-cell infusion after TCR alpha/beta depleted unrelated and haploidentical transplantation: Results of a pilot trial. Bone Marrow Transplant. 53:264–273.
Matsura T. 2014. Oxidized phosphatidylserine: Production and bioactivities. Yonago Acta Med. 57:119–127.
Migliavacca M., Assanelli A., Ferrua F., Cicalese M.P., Biffi A., Frittoli M., Silvani P., Chidini G., Calderini E., Mandelli A., Camporesi A., Milani R., Farinelli G., Nicoletti R., Ciceri F., Aiuti A., and Bernardo M.E. 2016. Pioglitazone as a novel therapeutic approach in chronic granulomatous disease. J. Allergy Clin. Immunol. 137:1913–1915.e2.
Morillo-Gutierrez B., Beier R., Rao K., Burroughs L., Schulz A., Ewins A.M., Gibson B., Sedlacek P., Krol L., Strahm B., Zaidman I., Kalwak K., Talano J.A., Woolfrey A., Fraser C., Meyts I., Müller I., Wachowiak J., Bernardo M.E., Veys P., Sykora K.W., Gennery A.R., and Slatter M. 2016. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: A multicenter experience. Blood. 128:440–448.
Nakazawa Y., Kawai T., Arai K., Tamura E., Uchiyama T., and Onodera M. 2017. Fecal calprotectin rise in chronic granulomatous disease-associated colitis. J. Clin. Immunol. 37:741–743.
Noel N., Mahlaoui N., Blanche S., Suarez F., Coignard-Biehler H., Durieu I., Godeberge P., Sokol H, Catherinot E., Poiree S., Chapdelaine H., Dunogue B., Bodemer C., Lecuit M., Fischer A., Lortholary O., and Hermine O. 2013. Efficacy and safety of thalidomide in patients with inflammatory manifestations of chronic granulomatous disease: A retrospective case series. J. Allergy Clin. Immunol. 132:997–1000.e4.
Parta M., Hilligoss D., Kelly C., Kwatemaa N., Theobald N., Malech H., and Kang E.M. 2015. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: A first report. J. Clin. Immunol. 35:675–680.
Reeves E.P., Lu H., Jacobs H.L., Messina C.G., Bolsover S., Gabella G., Potma E.O., Warley A., Roes J., and Segal A.W. 2002. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 416:291–297.
Regueiro-García A., Fariña-Nogueira S., Porto-Arceo J.Á., and Couselo-Sánchez J.M. 2018. Haploidentical stem cell transplantation in a boy with chronic granulomatous disease. Allergol. Immunopathol. 46:385–388.
Roos, D., Tool, A.T.J., van Leeuwen, K., and de Boer, M. 2017. Biochemical and genetic diagnosis of chronic granulomatous disease. In Chronic granulomatous disease: Genetics, biology and clinical management. Edited by R.A. Seger, D. Roos, B.H. Segal, and T.W. Kuijpers. New York, NY, USA: Nova Biomedical Press. pp. 231–300.
Sanford A.N., Suriano A.R., Herche D., Dietzmann K., and Sullivan K.E. 2006. Abnormal apoptosis in chronic granulomatous disease and autoantibody production characteristic of lupus. Rheumatology. 45:178–181.
Santilli G., Almarza E., Brendel C., Choi U., Beilin C., Blundell M.P., Haria S., Parsley K.L., Kinnon C., Malech H.L., Bueren J.A., Grez M., and Thrasher A.J. 2011. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol. Ther. 19:122–132.
Seger R.A. 2010. Hematopoietic stem cell transplantation for chronic granulomatous disease. Immunol. Allergy Clin. North Am. 30:195–208.
Seger, R.A., Roos, D., Segal, B.H., and Kuijpers, T.W. (Editors). 2017. Chronic granulomatous disease: Genetics, biology, clinical management [Internet]. New York, NY, USA: Nova Publishers.
Shah R.M., Elfeky R., Nademi Z., Qasim W., Amrolia P., Chiesa R., Rao K., Lucchini G., Silva J.M.F., Worth A., Barge D., Ryan D., Conn J., Cant A.J., Skinner R., Abd Hamid I.J., Flood T., Abinun M., Hambleton S., Gennery A.R., Veys P., and Slatter M. 2018. T-cell receptor αβ+ and CD19+ cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J. Allergy Clin. Immunol. 141:1417–1426.e1.
Siddiqui S., Anderson V.L., Hilligoss D.M., Abinun M., Kuijpers T.W., Masur H., Witebsky F.G., Shea Y.R., Gallin J.I., Malech H.L., and Holland S.M. 2007. Fulminant mulch pneumonitis: An emergency presentation of chronic granulomatous disease. Clin. Infect. Dis. 45:673–681.
Sirinavin S., Techasaensiri C., Benjaponpitak S., Pornkul R., and Vorachit M. 2005. Invasive Chromobacterium violaceum infection in children: Case report and review. Pediatr. Infect. Dis. J. 24:559–561.
Sollberger G., Tilley D.O., and Zychlinsky A. 2018. Neutrophil extracellular traps: The biology of chromatin externalization. Dev. Cell. 44:542–553.
Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H., Koehl U., Preiss C., Ball C., Martin H., Göhring G., Schwarzwaelder K., Hofmann W.K., Karakaya K., Tchatchou S., Yang R., Reinecke P., Kühlcke K., Schlegelberger B., Thrasher A.J., Hoelzer D., Seger R., von Kalle C., and Grez M. 2010. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 16:198–204.
Straughan D.M., McLoughlin K.C., Mullinax J.E., Marciano B.E., Freeman A.F., Anderson V.L., Uzel G., Azoury S.C., Sorber R., Quadri H.S., Malech H.L., DeRavin S.S., Kamal N., Koh C., Zerbe C.S., Kuhns DB, Gallin J.I., Heller T., Holland S.M., and Rudloff U. 2018. The changing paradigm of management of liver abscesses in chronic granulomatous disease. Clin. Infect. Dis. 66:1427–1434.
The International Chronic Granulomatous Disease Cooperative Study Group. 1991. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N. Engl. J. Med. 324:509–516.
Thomas D.C. 2017. The phagocyte respiratory burst: Historical perspectives and recent advances. Immunol. Lett. 192:88–96.
Thomas D.C., Clare S., Sowerby J.M., Pardo M., Juss J.K., Goulding D.A., van der Weyden L., Storisteanu D., Prakash A., Espéli M., Flint S., Lee J.C., Hoenderdos K., Kane L., Harcourt K., Mukhopadhyay S., Umrania Y., Antrobus R., Nathan J.A., Adams D.J., Bateman A., Choudhary J.S., Lyons P.A., Condliffe A.M., Chilvers E.R., Dougan G., and Smith K.G. 2017. Eros is a novel transmembrane protein that controls the phagocyte respiratory burst and is essential for innate immunity. J. Exp. Med. 214:1111–1128.
Thomas D.C., Charbonnier L.M., Schejtman A., Aldhekri H., Coomber E.L., Dufficy E.R., Beenken A.E., Lee J.C., Clare S., Speak A.O., Thrasher A.J., Santilli G., Al-Mousa H., Alkuraya F.S., Chatila T.A., and Smith K.G.C. 2019. EROS/CYBC1 mutations: Decreased NADPH oxidase function and chronic granulomatous disease. J. Allergy Clin. Immunol. 143:782–785.
Thomsen I.P., Smith M.A., Holland S.M., and Creech C.B. 2016. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J. Allergy Clin. Immunol. Pract. 4:1082–1088.
Thrasher, A. 2018. Gene therapy for X-linked chronic granulomatous disease. In 18th Biennial Meeting of the European Society for Immunodeficiencies, Lisbon, Portugal, 24–27 October. Abstract ESID 8-0108.
Uzel G., Orange J.S., Poliak N., Marciano B.E., Heller T., and Holland S.M. 2010. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin. Infect. Dis. 51:1429–1434.
van de Geer A., Nieto-Patlán A., Kuhns D.B., Tool A.T., Arias A.A., Bouaziz M., de Boer M., Franco J.L., Gazendam R.P., van Hamme J.L., van Houdt M., van Leeuwen K., Verkuijlen P.J., van den Berg T.K., Alzate J.F., Arango-Franco C.A., Batura V., Bernasconi A.R., Boardman B., Booth C., Burns S.O., Cabarcas F., Bensussan N.C., Charbit-Henrion F., Corveleyn A., Deswarte C., Azcoiti M.E., Foell D., Gallin J.I., Garcés C., Guedes M., Hinze C.H., Holland S.M., Hughes S.M., Ibañez P., Malech H.L., Meyts I., Moncada-Velez M., Moriya K., Neves E., Oleastro M., Perez L., Rattina V., Oleaga-Quintas C., Warner N., Muise A.M., López J.S., Trindade E., Vasconcelos J., Vermeire S., Wittkowski H., Worth A., Abel L., Dinauer M.C., Arkwright P.D., Roos D., Casanova J.L., Kuijpers T.W., and Bustamante J. 2018. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J. Clin. Invest. 128:3957–3975.
van den Berg J.M., van Koppen E., Åhlin A., Belohradsky B.H., Bernatowska E., Corbeel L., Español T., Fischer A., Kurenko-Deptuch M., Mouy R., Petropoulou T., Roesler J., Seger R., Stasia M.J., Valerius N.H., Weening R.S., Wolach B., Roos D., and Kuijpers T.W. 2009. Chronic granulomatous disease: The European experience. PLoS ONE. 4:e5234.
Winkelstein J.A., Marino M.C., Johnston R.B. Jr., Boyle J., Curnutte J., Gallin J.I., Malech H.L., Holland S.M., Ochs H., Quie P., Buckley R.H., Foster C.B., Chanock S.J., and Dickler H. 2000. Chronic granulomatous disease: Report on a national registry of 368 patients. Medicine. 79:155–169.
Wolach B., Gavrieli R., de Boer M., van Leeuwen K., Berger-Achituv S., Stauber T., Ben Ari J., Rottem M., Schlesinger Y., Grisaru-Soen G., Abuzaitoun O., Marcus N., Zion Garty B., Broides A., Levy J., Stepansky P., Etzioni A., Somech R., and Roos D. 2017. Chronic granulomatous disease: Clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am. J. Hematol. 92:28–36.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 6 • Number 1 • March 2019
Pages: 1 - 16
History
Received: 31 August 2018
Accepted: 15 October 2018
Accepted manuscript online: 28 January 2019
Copyright
© 2019.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
Reinhard A.Seger. 2019. Chronic granulomatous disease 2018: advances in pathophysiology and clinical management. LymphoSign Journal.
6(1): 1-16. https://doi.org/10.14785/lymphosign-2018-0012
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
