Abstracts from the Immunodeficiency Canada—5th SCID Symposium, Toronto, ON, 12 October 2017
Severe combined immunodeficiency with microcephaly, growth retardation, and sensitivity to ionizing radiation is caused by mutation in the NHEJ1 gene
Hannah Roberts, Stuart Turvey, Victoria Cook, Kyla Hildebrand
Division of Allergy and Clinical Immunology, Department of Pediatrics, University of British Columbia, BC Children’s Hospital, Vancouver, BC, Canada
Background: Severe combined immunodeficiency (SCID) with microcephaly, growth retardation, and sensitivity to ionizing radiation is a novel autosomal recessive primary immunodeficiency (OMIM #611291). This form of SCID is caused by autosomal recessive mutations in the non-homologous end-joining factor 1 (NHEJ1) gene on chromosome 2q35, which encodes cernunnos protein. Since this syndrome was first described in 2006, only 5 patients with a similar phenotype have been reported from 4 unrelated families.
The NHEJ1 gene is a repair gene in the non-homologous end-joining (NHEJ) pathway. The NHEJ pathway repairs DNA double strand breaks. Failure of this pathway can have deleterious results and can impact development, genetic stability, and cause immunodeficiency. NHEJ1 is primarily expressed in the cerebellum and cerebral cortex, and may play a role in brain development (Figure 1).
Figure 1:
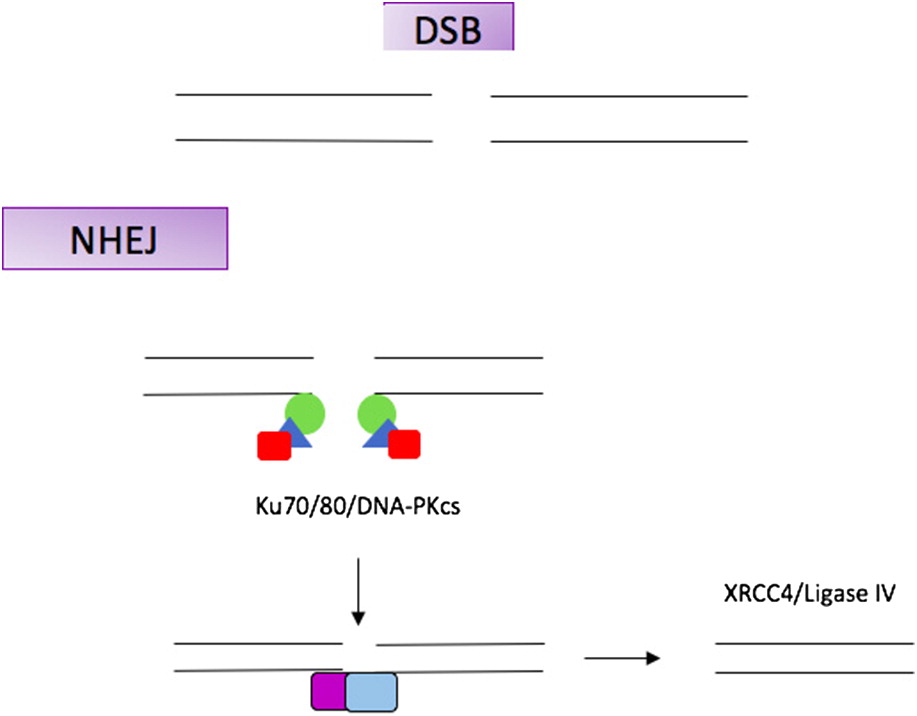
We report a 20-month-old female who was found to have an autosomal recessive compound heterozygous mutation in NHEJ1. Two novel pathogenic variants in NHEJ1 were identified, including c.350delT and c.355T>G. The c.350delT mutation causes a frameshift, replacing the phenylalanine residue at codon position 117 to a serine residue, ultimately leading to a premature stop codon at position 9 of the new reading frame (p.F117SfsX9). This pathogenic variant is predicted to affect normal protein function. The c.355T>G mutation substitutes the amino acid tryptophan for glycine at position 119 (p.W119G) and is predicted to damage protein structure. Our patient’s specific mutation is novel, but her clinical presentation is consistent with the phenotypic features found in the very small patient population with NHEJ1 mutations.
The clinical presentation and disease trajectory of NHEJ1-SCID is similar in the population of patients reported. To date, all 5 patients with this form of SCID had failure to thrive, microcephaly, and recurrent infections. Two unrelated individuals died of septic shock at age 18 and 4 years old. All patients had severe T and B cell lymphocytopenia and hypogammaglobinemia, with low IgG, IgA, but normal or increased IgM. Natural Killer cell numbers were normal in all 5 individuals. The patients’ fibroblasts showed elevated sensitivity to ionizing radiation. Like our patient, 2 individuals had autoimmune features including autoimmune anemia and thrombocytopenia. Ahnesorge et al. (2006) identified a mutation in the NHEJ1 gene, in a patient previously reported in 2003 to have SCID and increased sensitivity to ionizing radiation, but without microcephaly.
As patients with NHEJ1-SCID have been shown to have severe infections, with potential for mortality, proper management and follow-up is crucial. Management strategies for other forms of SCID include IgG substitution, antibiotics, immune modulation, and hematopoietic stem cell transplantation; however, evidence-based strategies for this novel form of SCID are lacking. Sensitivity to ionizing radiation is a prominent feature and adds complexity to patient management (Dai et al. 2003; Ahnesorg et al. 2006; Buck et al. 2006; Callebaut et al. 2006; Cantagrel et al. 2007).
Methods: The patient’s initial immunology assessment was at age 15 months. During this time, our patient was evaluated for a genetic cause with whole exome sequencing. The results were diagnostic of SCID caused by 2 novel pathogenic variants in the NEHJ1 gene, with autosomal recessive inheritance. As this is a recent diagnosis, the patient is currently being followed by Immunology and the Bone Marrow Transplant team.
Results:
Clinical features: This is a now 20-month-old female who was born at term weighing 2.4 kg. She had a history of intrauterine growth retardation and failure to thrive. Antenatal ultrasounds documented microcephaly. Amniocentesis was unremarkable and chromosomal microarray was normal. She was initially assessed at 15 months by Immunology, prior to diagnosis of SCID, during hospitalization for autoimmune hemolytic anemia. On admission to hospital, head circumference, height, and weight were >2 standard deviations below the mean for age. On initial consultation, she had no unusual or recurrent infections. She had several mild, self-resolving viral illnesses each lasting 2–3 days. She had no clinical history of sinopulmonary infections and had never received antibiotics. Her vaccinations were up to date per provincial schedule and she tolerated live vaccines, including rotavirus, varicella, and MMR without adverse reaction. Her umbilical cord fell off within 1 week. She met developmental milestones as expected. She is the first and only child to non-consanguineous parents. The father is of Middle Eastern descent and the mother is Chinese. There was no pertinent family history.
Following her diagnosis, she has been followed jointly by Immunology and the Bone Marrow Transplant team. She is currently receiving intravenous immunoglobulin monthly, Trimethoprim/Sulfamethoxazole prophylaxis, and Mycophenolate Mofetil (MMF) for autoimmune cytopenias. She has remained infection free. Growth continues to be proportional and under the third percentile for age. Development continues to be appropriate. Options for hematopoietic stem cell transplantation are currently under discussion.
Immunological features: When first assessed at 15 months, CBC revealed a white blood cell count of 6.8, neutrophils were low at 0.84, and lymphocytes normal at 4.99. Hemoglobin was low at 76, and platelets normal at 314. Serum immunoglobulin levels were abnormal: IgG was very low at 0.3 g/L, IgA undetectable, and IgM was normal at 1.07 g/L.
T and B lymphocyte subsets were abnormal: CD3 normal, CD8 normal, CD4 low at 0.63, CD19 low at 0.28. CD4 to CD8 ratio reduced at 0.41, and NK cells normal at 0.59.
Mitogen stimulations were within normal limits:
1.
PHA—proliferation count of 7541 (4484–37 943)
2.
PWM—proliferation count of 1508 (422–21 922)
3.
SAC—proliferation count 1187 (413–14 027)
Conclusions: We report a 20-month-old female with an autosomal recessive NHEJ1 (c.350delT and c.355T>G) gene mutation. To our knowledge, there are 5 patients reported with this rare condition. Our patient’s features of profound T and B cell lymphocytopenia, hypogammaglobinemia, autoimmune hemolytic anemia, and microcephaly are consistent with those described in other patients with NHEJ1-SCID. Our patient’s clinical presentation is unique, however, as she has been free of recurrent or unusual infections, and her development has been normal. Out of the very small patient population with NHEJ1-SCID, 2 patients have died as a result of septic shock. Due to the high risk of mortality, hematopoietic stem cell transplantation is an important treatment consideration. In this unique patient population, the process is complicated by sensitivity to ionizing radiation, propensity for chromosomal breakage secondary to ineffective cernunnos, and finding an appropriate donor. As this is a very rare and novel condition, reporting our patient is beneficial to the Immunology community. Further research is needed to fully understand the impact of defective cernunnos on brain development and disease trajectory.
REFERENCES
Ahnesorg, P., Smith, P., and Jackson, S.P. 2006. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 124:301–313. PMID: https://pubmed.ncbi.nlm.nih.gov/16439205/. doi: https://doi.org/10.1016/j.cell.2005.12.031.
Buck, D., Malivert, L., de Chasseval, R., Barraud, A., Fondanèche, M.C., Sanal, O., Plebani, A., Stéphan, J.L., Hufnagel, M., le Deist, F., Fischer, A., Durandy, A., de Villartay, J.P., and Revy, P. 2006. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 124:287–299. PMID: https://pubmed.ncbi.nlm.nih.gov/16439204/. doi: https://doi.org/10.1016/j.cell.2005.12.030.
Callebaut, I., Malivert, L., Fischer, A., Mornon, J.P., Revy, P., and de Villartay, J.P. 2006. Cernunnos interacts with the XRCC4·DNA-ligase IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1. J. Biol. Chem. 281:13857–13860. PMID: https://pubmed.ncbi.nlm.nih.gov/16571728/. doi: https://doi.org/10.1074/jbc.C500473200.
Cantagrel, V., Lossi, A.M., Lisgo, S., Missirian, C., Borges, A., Philip, N., Fernandez, C., Cardoso, C., Figarella-Branger, D., Moncla, A., Lindsay, S., Dobyns, W.B., and Villard, L. 2007. Truncation of NHEJ1 in a patient with polymicrogyria. Hum. Mutat. 28(4):356–364. PMID: https://pubmed.ncbi.nlm.nih.gov/17191205/. doi: https://doi.org/10.1002/humu.20450.
Dai, Y., Kysela, B., Hanakahi, L.A., Manolis, K., Riballo, E., Stumm, M., Harville, T.O., West, S.C., Oettinger, M.A., and Jeggo, P.A. 2003. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc. Natl. Acad. Sci. U.S.A. 100:2462–2467. PMID: https://pubmed.ncbi.nlm.nih.gov/12604777/. doi: https://doi.org/10.1073/pnas.0437964100.
Davis, A.J., and Chen, D.J. 2013. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2(3):130–143. PMID: https://pubmed.ncbi.nlm.nih.gov/24000320/. doi: https://doi.org/10.3978/j.issn.2218-676X.2013.04.02.
Maintenance of T cell receptor excision circles (TREC) levels in zeta chain-associated protein kinase of 70 kD (ZAP70) deficiency
Sneha Suresha, Chaim M. Roifmana,b
aDepartment of Immunology and Allergy, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada; bThe Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children, Toronto, ON, Canada
Background: ZAP70 is a tyrosine kinase whose primary role is in signaling downstream of the T cell receptor (Arpaia et al. 1994). It is activated after recruitment to phosphorylated motifs on CD3ζ and phosphorylation by Lck, a Src family kinase (Wange et al. 1995). Once activated, ZAP70 phosphorylates numerous proteins including LAT (linker of activated T cells), SLP-76 and Cbl (Roifman et al. 2010). The downstream actions of ZAP70 are multipronged—it helps mediate intracellular calcium signaling, helps maintain the cytoskeleton framework of the immunologic synapse, as well as having roles in T cell development and regulation (Arpaia et al. 1994; Au-Yeung et al. 2009). Clinical manifestations of ZAP70 deficiency include CD8 lymphopenia, impaired T cell function and severe combined immunodeficiency, with varying degrees of autoimmunity (Arpaia et al. 1994; Shirkani et al. 2017).
As CD4 T cell numbers are preserved in ZAP70 deficiency, and the surmised primary role of ZAP70 is downstream to T cell receptor (TCR) development, it was plausible to assume that TREC levels—a byproduct of T cell receptor development, would be normal. However, previous studies have shown that TREC levels in ZAP70 deficiency may actually be low in patients diagnosed after 6 months of age, suggesting that ZAP70 has an important role in pre-T cell receptor development (Roifman et al. 2010, 2012; Liu et al. 2017). Yet, this observation was not always consistent. We report here 4 cases of ZAP70 deficiency that had TREC levels above established newborn screening cutoffs for severe combined immunodeficiency (SCID), but gradually declined in a variable rate.
Methods: Consent was obtained for a retrospective case series of 4 patients with ZAP70 deficiency, as well as research testing on clinical samples. Whole blood and dried blood spot TREC levels were measured via real-time polymerase chain reaction (PCR).
Results:
Clinical manifestations: Patient 1 was diagnosed after a history of chronic diarrhea, recurrent lower respiratory tract infections, and a respiratory deterioration at 7 months of age requiring extra-corporeal life support. A bronchoscopy was positive for multiple organisms including Pneumocystis jirovecii, Parainfluenza type 3, Candida albicans, and Klebsiella pneumoniae. He was given an emergency, unconditioned 10/10 matched sibling donor hematopoietic stem cell transplant (HSCT) (Kim et al. 2013), and then eventual re-transplant after engraftment issues. Patient 2, patient 1’s sibling, was diagnosed postnatally, and had prompt institution of isolation precautions and antimicrobial prophylaxis, with eventual HSCT at 5 months of age. Patient 3 was relatively thriving until respiratory failure at 5 months of age, with bronchoscopy positive for Pneumocystis jirovecii, parainfluenza type 1, and Enterobacter sp. He received a 10/10 matched sibling donor HSCT at 5 months of age. Patient 4 was diagnosed at 9 months of age, after a history of refractory diaper dermatitis, oral thrush, and recurrent lower respiratory tract infections with pathogens including RSV and bocavirus. He received at 10/10 matched unrelated transplant at 11 months of age.
Laboratory investigations: Results of laboratory investigations are shown in Table 1. All patients had mutation IVS12-11G>A, c.[1624-11G>A]; c.[1624-11G>A] in ZAP70. Newborn screen (NBS) TREC levels were done at 0–7 days of life while all whole blood TREC levels were done periodically up to HSCT. Local cutoff values for abnormal newborn screening are flagged for those <75 copies/3 μL DNA. All patients had NBS levels far exceeding this level (van der Spek et al. 2015). Over a course of up to 1 year, TREC levels declined and some reached abnormal levels.
Table 1:
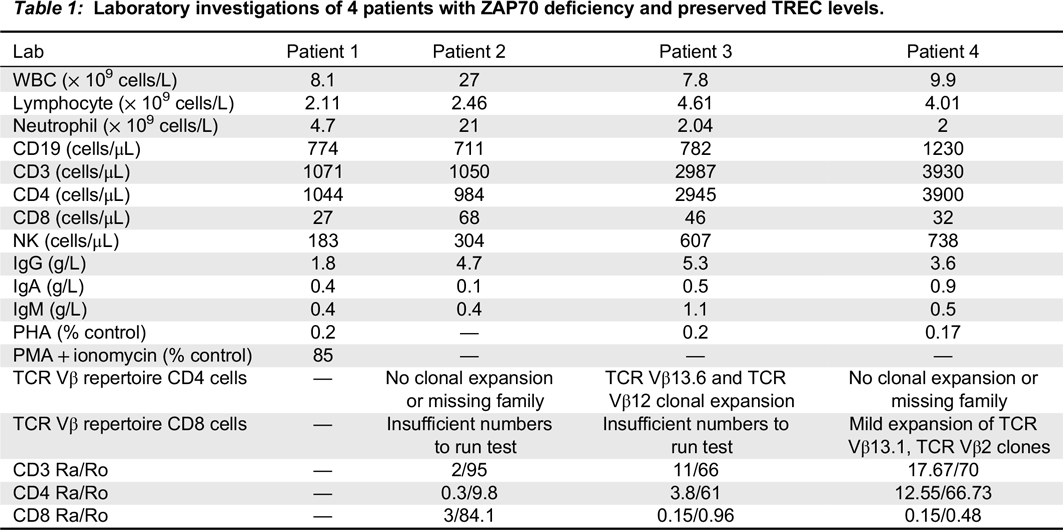
Conclusions: We report here 4 cases of ZAP70 deficiency with sustained TREC levels at birth and in the first year of life. Both NBS and subsequent whole blood TREC levels in patients with ZAP70 deficiency are in the normal range at birth and in young infants, but tend to decline over time. This indicates that thymopoiesis in ZAP70 deficiency is not durable. This finding has important implications for both newborn screening programs for SCID, as well as the pathophysiology of ZAP70 deficiency.
REFERENCES
Arpaia, E., Shahar, M., Dadi, H., Cohen, A., and Roifman, C.M. 1994. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking ZAP-70 kinase. Cell. 76:947–958. PMID: https://pubmed.ncbi.nlm.nih.gov/8124727/. doi: https://doi.org/10.1016/0092-8674(94)90368-9.
Au-Yeung, B.B., Deindl, S., Hsu, L.Y., Palacios, E.H., Levin, S.E., Kuriyan, J., and Weiss, A. 2009. The structure, regulation, and function of ZAP-70. Immunol. Rev. 228:41–57. PMID: https://pubmed.ncbi.nlm.nih.gov/19290920/. doi: https://doi.org/10.1111/j.1600-065X.2008.00753.x.
Kim, V.H.-D., Murguia, L., Schechter, T., Grunebaum, E., and Roifman, C.M. 2013. Emergency treatment for ζ chain-associated protein of 70 kDa (ZAP70) deficiency. J. Allergy Clin. Immunol. 131(4):1233–1235. PMID: https://pubmed.ncbi.nlm.nih.gov/23141738/. doi: https://doi.org/10.1016/j.jaci.2012.09.020.
Liu, Q., Wang, Y.P., Liu, Q., Zhao, Q., Chen, X.M., Xue, X.H., Zhou, L.N., Ding, Y., Tang, X.M., Zhao, X.D., and Zhang, Z.Y. 2017. Novel compound heterozygous mutations in ZAP70 in a Chinese patient with leaky severe combined immunodeficiency disorder. Immunogenetics. 69(4):199–209. PMID: https://pubmed.ncbi.nlm.nih.gov/28124082/. doi: https://doi.org/10.1007/s00251-017-0971-0.
Roifman, C.M., Dadi, H., Somech, R., Nahum, A., and Sharfe, N. 2010. Characterization of ζ-associated protein, 70 kd (ZAP70)-deficient human lymphocytes. J. Allergy Clin. Immunol. 126:1226–1233.e1. PMID: https://pubmed.ncbi.nlm.nih.gov/20864151/. doi: https://doi.org/10.1016/j.jaci.2010.07.029.
Roifman, C.M., Somech, R., Kavadas, F., Pires, L., Nahum, A., Dalal, I., and Grunebaum, E. 2012. Defining combined immunodeficiency. J. Allergy Clin. Immunol. 130(1):177–183. PMID: https://pubmed.ncbi.nlm.nih.gov/22664165/. doi: https://doi.org/10.1016/j.jaci.2012.04.029.
Shirkani, A., Shahrooei, M., Azizi, G., Rokni-Zadeh, H., Abolhassani, H., Farrokhi, S., Frans, G., Bossuyt, X., and Aghamohammadi, A. 2017. Novel mutation of ZAP-70-related combined immunodeficiency: First case from the national Iranian registry and review of the literature. Immunol. Invest. 46(1):70–79. PMID: https://pubmed.ncbi.nlm.nih.gov/27759478/. doi: https://doi.org/10.1080/08820139.2016.1214962.
van der Spek, J., Groenwold, R.H., van der Burg, M., and van Montfrans, J.M. 2015. TREC based newborn screening for severe combined immunodeficiency disease: A systematic review. J. Clin. Immunol. 35:416–430. PMID: https://pubmed.ncbi.nlm.nih.gov/25893636/. doi: https://doi.org/10.1007/s10875-015-0152-6.
Wange, R.L., Guitián, R., Isakov, N., Watts, J.D., Aebersold, R., and Samelson, L.E. 1995. Activating and inhibitory mutations in adjacent tyrosines in the kinase domain of ZAP-70. J. Biol. Chem. 270:18730–18733. PMID: https://pubmed.ncbi.nlm.nih.gov/7642520/. doi: https://doi.org/10.1074/jbc.270.32.18730.
Moesin deficiency: on the spectrum of combined immunodeficiency
Amarilla Mandolaa,b, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, Toronto, ON, Canada; bCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
Background: Ezrin, radixin, and moesin (ERM) proteins are structural components of the intracellular cortex and are expressed in a variety of cell types. The function of these proteins is to maintain cell shape, microvilli formation, organize cell membranes for migration (pseudopod/uropod formation), immune synapse (IS) formation, phagocytosis, and apoptosis (Niggli and Rossy 2008). The activation of ERM proteins by phosphorylation leads to intramolecular conformational changes which unmasks sites for interaction with other proteins critically involved in shape regulation—including actin filaments, transmembrane proteins (membrane PIP, L-selectin, CD43(IS), CD44, and ICAM-1), and scaffolding proteins (EBP50 for CFTR, PDGFR, β2AR). Dysregulation of ERM proteins, particularly moesin, disrupts human lymphoid and neutrophil cells (Niggli and Rossy 2008; Liu et al. 2015; Lagresle-Peyrou et al. 2016). Moesin is ubiquitously expressed in lungs, spleen, kidney, endothelial cells of vessels, and is the predominant ERM protein in lymphocytes and neutrophils (Liu et al. 2015). Given the role and unique expression of this ERM protein, it is clear that moesin has important non-redundant roles in immune function.
Methods: We report on a patient who was evaluated in the IDEA Complex Immunology Clinic at The Hospital for Sick Children, and was enrolled in the Primary Immunodeficiency Registry and Tissue Bank under REB protocol number 1000005598.
Results: Our patient is a 54-year-old male, from a non-consanguineous family, who presented with 3 major invasive bacterial infections: bacterial meningitis at age 5 years, pneumonia, ARDS and septic shock at age 46 years (the pathogen was not identified), as well as cellulitis of the face due to Staphylococcal infection at age 50 years. He suffers from recurrent oral ulcers, recurrent sinusitis, and recurrent leg cellulitis with ulceration. He has a family history of a large amount of infections and a maternal uncle who died from Legionella sepsis at age 40 years. His immune evaluation revealed chronic anemia, profound lymphopenia, abnormal flow cytometry with low CD3+ T cells including both CD4+ and CD8+, as well as low CD19+ and NK cells. He had abnormal phytohemagglutinin response. He was found to have normal albumin as well as immunoglobulin levels (IgG was normal low at 7.8), reactive specific antibody titers, NOBI, CH50, and chromosomal microarray as well. Genetic analysis revealed a novel mutation in the moesin gene (MSN).
Discussion: Moesin deficiency has only recently been described and this is the second report. Patients with moesin deficiency can present during infancy or childhood with a severe form of the disease or with a milder form during adulthood (Lagresle-Peyrou et al. 2016). The clinical manifestation was consistent with the sole previous report.
REFERENCES
Lagresle-Peyrou, C., Luce, S., Ouchani, F., Soheili, T.S., Sadek, H., Chouteau, M., Durand, A., Pic, I., Majewski, J., Brouzes, C., Lambert, N., Bohineust, A., Verhoeyen, E., Cosset, F.L., Magerus-Chatinet, A., Rieux-Laucat, F., Gandemer, V., Monnier, D., Heijmans, C., van Gijn, M., Dalm, V.A., Mahlaoui, N., Stephan, J.L., Picard, C., Durandy, A., Kracker, S., Hivroz, C., Jabado, N., de Saint Basile, G., Fischer, A., Cavazzana, M., and André-Schmutz, I. 2016. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J. Allergy Clin. Immunol. 138:1681–1689.e8. PMID: https://pubmed.ncbi.nlm.nih.gov/27405666/. doi: https://doi.org/10.1016/j.jaci.2016.04.032.
Liu, X., Yang, T., Suzuki, K., Tsukita, S., Ishii, M., Zhou, S., Wang, G., Cao, L., Qian, F., Taylor, S., Oh, M.J., Levitan, I., Ye, R.D., Carnegie, G.K., Zhao, Y., Malik, A.B., and Xu, J. 2015. Moesin and myosin phosphatase confine neutrophil orientation in a chemotactic gradient. J. Exp. Med. 212:267–280. PMID: https://pubmed.ncbi.nlm.nih.gov/25601651/. doi: https://doi.org/10.1084/jem.20140508.
Niggli, V., and Rossy, J. 2008. Ezrin/radixin/moesin: Versatile controllers of signaling molecules and of the cortical cytoskeleton. Int. J. Biochem. Cell Biol. 40:344–349. PMID: https://pubmed.ncbi.nlm.nih.gov/17419089/. doi: https://doi.org/10.1016/j.biocel.2007.02.012.
Newborn screening for SCID detects infants with no primary immunodeficiency
Mohammad Alsalamaha, Sneha Suresha, Brenda Reida, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
Background: Severe combined immunodeficiency (SCID) is a life-threatening disease with an asymptomatic period and a curative treatment that is time-sensitive (Pai et al. 2014). Newborn Screening (NBS) for SCID was first introduced as a pilot study in 2008, using the measurement of T cell receptor excision circles (TRECs) as an index of T cell receptor development (Kwan et al. 2014). This test is inexpensive and can be readily performed on Guthrie cards, with acceptable sensitivity and specificity (Chan and Puck 2005; McGhee et al. 2005; Morinishi et al. 2009). For these reasons, SCID met the criteria to be a candidate for NBS (Wilson and Jungner 1968).
After the initial success of NBS in Wisconsin, many states in the USA also implemented SCID NBS (Kwan et al. 2014). In 2013, Immunodeficiency Canada spearheaded the campaign to introduce SCID NBS in Ontario, as part of the provincial NBS program. To date, the program has been successful in identifying otherwise undiagnosed patients with SCID. However, as with most screening tests, there is potential for both false-negative and false-positive results.
Causes of false-negative results include primary immunodeficiency disease (PID) associated with significant residual autologous T cells (combined immunodeficiency). These conditions may not have critical lymphopenia at birth, have dysfunctional T cells with only a missing subset (such as MHC class I or II), or those affecting T cell maturation after the VDJ recombination process (ZAP70). False-negative results have been discussed in the past, with some approaches suggested that may improve the screening program which still remains inherently devoid of solutions (Roifman et al. 2012; Grazioli et al. 2014).
True false-positive screen tests consist of positive screens which could not subsequently be confirmed by a second TREC test. This can occur because of laboratory error or inadequate sample. These causes are out of the scope of this report. Approaches to increase the sensitivity of TREC-based NBS may result in increased detection of non-SCID cases. This may bring about undue stress for families as well as increase healthcare utilization (Gurian et al. 2006).
Here, we report our 4-year experience in the largest quaternary referral centre with the identification of NBS-positive cases that turned out to have no evidence of PID.
Methods: We retrospectively analyzed all SCID NBS-positive results that were evaluated at The Hospital for Sick Children, Ontario. TREC levels were measured via polymerase chain reaction on Guthrie spot cards. Positive newborn screens were defined as those with undetectable TREC levels. In 2014, the threshold was changed to those with TRECs <75 copies/3 μL DNA.
Results: Since August 2013, 63 patients have been evaluated at The Hospital for Sick Children for SCID NBS-positive results. Five of the 63 patients were subsequently diagnosed with SCID (1 patient with ADA deficiency, 1 patient with Coronin 1A deficiency, 1 with classic SCID, and 2 patients with Omenn’s Syndrome with unknown genetic mutation). Four of the 63 had non-SCID PID (2 patients with a mutation in 22q11, 1 patient with Ataxia-Telangiectasia, and 1 with pending genetic work-up). The remaining 54 infants were deemed to have no PID. Details of these patients are outlined in Table 1.
Table 1:

a
Some patients had more than 1 likely cause and were tallied more than once.
b
Perinatal stress is defined as abnormal fetal monitoring requiring intervention, respiratory distress at birth, shock, encephalopathy, congenital diaphragmatic hernia, IUGR, necrotizing enterocolitis, hypoglycemia requiring intervention, fetal hydrops, twin-twin transfusion, or cardiac conditions.
c
Maternal factors are defined as those receiving immunosuppressive medications and confirmed active infection (by microbiological work-up) during delivery.
Discussion: We present here multiple causes for NBS-positive results. The main causes in our centre are prematurity, use of immunosuppressive agents in mother or infant, incidental thymectomy for cardiac surgery, and acute stress (Table 1). In some cases causes were not identified.
Conclusions: This evidence is of great clinical importance as it provides physicians and genetic counselors with the data that most NBS-positive cases are detected in infants with no PID.
REFERENCES
Chan, K., and Puck, J.M. 2005. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 115(2):391–398. PMID: https://pubmed.ncbi.nlm.nih.gov/15696101/. doi: https://doi.org/10.1016/j.jaci.2004.10.012.
Grazioli, S., Bennett, M., Hildebrand, K.J., Vallance, H., Turvey, S.E., and Junker, A.K. 2014. Limitation of TREC-based newborn screening for ZAP70 severe combined immunodeficiency. Clin. Immunol. 153(1):209–210. PMID: https://pubmed.ncbi.nlm.nih.gov/24797280/. doi: https://doi.org/10.1016/j.clim.2014.04.015.
Gurian, E.A., Kinnamon, D.D., Henry, J.J., and Waisbren, S.E. 2006. Expanded newborn screening for biochemical disorders: The effect of a false-positive result. Pediatrics. 117(6):1915–1921. PMID: https://pubmed.ncbi.nlm.nih.gov/16740831/. doi: https://doi.org/10.1542/peds.2005-2294.
Kwan, A., Abraham, R.S., Currier, R., Brower, A., Andruszewski, K., Abbott, J.K., Baker, M., Ballow, M., Bartoshesky, L.E., Bonilla, F.A., Brokopp, C., Brooks, E., Caggana, M., Celestin, J., Church, J.A., Comeau, A.M., Connelly, J.A., Cowan, M.J., Cunningham-Rundles, C., Dasu, T., Dave, N., De La Morena, M.T., Duffner, U., Fong, C.T., Forbes, L., Freedenberg, D., Gelfand, E.W., Hale, J.E., Hanson, I.C., Hay, B.N., Hu, D., Infante, A., Johnson, D., Kapoor, N., Kay, D.M., Kohn, D.B., Lee, R., Lehman, H., Lin, Z., Lorey, F., Abdel-Mageed, A., Manning, A., McGhee, S., Moore, T.B., Naides, S.J., Notarangelo, L.D., Orange, J.S., Pai, S.Y., Porteus, M., Rodriguez, R., Romberg, N., Routes, J., Ruehle, M., Rubenstein, A., Saavedra-Matiz, C.A., Scott, G., Scott, P.M., Secord, E., Seroogy, C., Shearer, W.T., Siegel, S., Silvers, S.K., Stiehm, E.R., Sugerman, R.W., Sullivan, J.L., Tanksley, S., Tierce, M.L. IV, Verbsky, J., Vogel, B., Walker, R., Walkovich, K., Walter, J.E., Wasserman, R.L., Watson, M.S., Weinberg, G.A., Weiner, L.B., Wood, H., Yates, A.B., Puck, J.M., and Bonagura, V.R. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 312(7):729–738. PMID: https://pubmed.ncbi.nlm.nih.gov/25138334/. doi: https://doi.org/10.1001/jama.2014.9132.
McGhee, S.A., Stiehm, E.R., and McCabe, E.R. 2005. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J. Pediatr. 147(5):603–608. PMID: https://pubmed.ncbi.nlm.nih.gov/16291349/. doi: https://doi.org/10.1016/j.jpeds.2005.06.001.
Morinishi, Y., Imai, K., Nakagawa, N., Sato, H., Horiuchi, K., Ohtsuka, Y., Kaneda, Y., Taga, T., Hisakawa, H., Miyaji, R., Endo, M., Oh-Ishi, T., Kamachi, Y., Akahane, K., Kobayashi, C., Tsuchida, M., Morio, T., Sasahara, Y., Kumaki, S., Ishigaki, K., Yoshida, M., Urabe, T., Kobayashi, N., Okimoto, Y., Reichenbach, J., Hashii, Y., Tsuji, Y., Kogawa, K., Yamaguchi, S., Kanegane, H., Miyawaki, T., Yamada, M., Ariga, T., and Nonoyama, S. 2009. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J. Pediatr. 155(6):829–833. PMID: https://pubmed.ncbi.nlm.nih.gov/19628217/. doi: https://doi.org/10.1016/j.jpeds.2009.05.026.
Pai, S.Y., Logan, B.R., Griffith, L.M., Buckley, R.H., Parrott, R.E., Dvorak, C.C., Kapoor, N, Hanson, I.C., Filipovich, A.H., Jyonouchi, S., Sullivan, K.E., Small, T.N., Burroughs, L., Skoda-Smith, S., Haight, A.E., Grizzle, A., Pulsipher, M.A., Chan, K.W., Fuleihan, R.L., Haddad, E., Loechelt, B., Aquino, V.M., Gillio, A., Davis, J., Knutsen, A., Smith, A.R., Moore, T.B., Schroeder, M.L., Goldman, F.D., Connelly, J.A., Porteus, M.H., Xiang, Q., Shearer, W.T., Fleisher, T.A., Kohn, D.B., Puck, J.M., Notarangelo, L.D., Cowan, M.J., and O’Reilly, R.J. 2014. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N. Engl. J. Med. 371(5):434–446. PMID: https://pubmed.ncbi.nlm.nih.gov/25075835/. doi: https://doi.org/10.1056/NEJMoa1401177.
Roifman, C.M., Somech, R., Kavadas, F., Pires, L., Nahum, A., Dalal, I., and Grunebaum E. 2012. Defining combined immunodeficiency. J. Allergy Clin. Immunol. 130(1):177–183. PMID: https://pubmed.ncbi.nlm.nih.gov/22664165/. doi: https://doi.org/10.1016/j.jaci.2012.04.029.
Wilson, J.M., and Jungner, Y.G. 1968. [Principles and practice of mass screening for disease]. Bol. Oficina Sanit. Panam. 65(4):281–393. PMID: https://pubmed.ncbi.nlm.nih.gov/4234760/.
Interleukin-2 receptor common gamma chain (IL2RG) defects present a diagnostic challenge
Caroline Weissera, Dennis E. Bulmanb, Kayla Flamenbaumc, Maian Roifmanc,d
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada; bCHEO Research Institute and Newborn Screening Ontario, Department of Pediatrics, University of Ottawa, Ottawa, ON, Canada; cThe Prenatal Diagnosis and Medical Genetics Program, Department of Obstetrics and Gynaecology, Mount Sinai Hospital, Toronto, ON, Canada; dDivision of Clinical and Metabolic Genetics, Department of Paediatrics, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
Background: Severe combined immunodeficiency (SCID) represents a rare group of primary immunodeficiency disorders (PID) characterized by a reduced number of T lymphocytes in association with a functional or quantitative defect in B lymphocytes, natural killer cells, or both. Patients with SCID may have known or yet unidentified genetic alterations explaining their phenotype. Mutations in the IL2RG gene, which encodes the common gamma chain of the interleukin-2 receptor, cause X-linked SCID (XL-SCID) as well as X-linked combined immunodeficiency and remain the most common cause of SCID. This protein is an important signaling component of many interleukin receptors, including those of interleukin-2, -4, -7, and -21, and has therefore been commonly known as the common gamma chain (Shearer et al. 2014).
Transplacental maternal engraftment (TME) is defined as the presence of maternal T cells in peripheral blood before bone marrow transplantation. The human placenta allows for bi-directional passage of nucleated cells between mother and fetus, and in healthy infants the immune system eradicates maternal cells. Patients with SCID lack the functional immunity required to reject circulating maternal T cells, resulting in persistent TME in up to 40% of these patients (Fischer et al. 1997; Liu et al. 2016). Although TME can be asymptomatic, some infants with SCID and TME can have clinical symptoms of graft-versus-host disease (GvHD) at diagnosis (Wahlstrom et al. 2017). TME has also been an impediment to proper infant immune evaluation as well as genetic analysis. Genetic testing is extremely important in SCID as early diagnosis allows for life-saving interventions such as bone marrow transplantation, which results in a higher survival rate when administered during the first 3 months of life (Kwan et al. 2014; Wahlstrom et al. 2015). In addition, proper molecular diagnosis aids in the important task of family genetic counseling.
Here, we present a patient with TME that posed a challenge to both genetic diagnosis as well as genetic counselling.
Methods:
Patient: Following informed consent, patient information was collected from medical records in accordance with REB Protocol No. 1000005598.
Sanger sequencing: Genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers. Sequencing was performed using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Next generation sequencing: Massively parallel sequencing was performed on a panel of 20 SCID genes (ADA, AK2, CARD11, CD247, CD3D, CD3E, DCLRE1C, IL2RG, IL7R, JAK3, LIG4, NHEJ1, PNP, PRKDC, PTPRC, RAC2, RAG1, RAG2, RMRP, ZAP70), following standard procedures for DNA sample preparation. Libraries were constructed using the Kapa Hyper Prep kit, and targeted capture of coding exons as well as splice junctions performed with Nimblegen’s SeqCap EZ Choice. Sequencing was done with 150 bp paired-end reads on an Illumina MiSeq.
Case Presentation: Our patient, now 25 years old, was diagnosed with SCID at 5 months of age with the presence of TME at time of diagnosis. He was one of the first patients world-wide to receive a matched unrelated bone marrow transplantation at 1 year of age and conditioning consisted of Busulfan and Cyclophosphamide. He had an uneventful transplant course with the exception of mild cutaneous GvHD. His engraftment was full and rapid with no complications. He continues to do extremely well 2 decades later, with no episodes of infections, autoimmunity or atopy. His engraftment remains solid and immune reconstitution is complete.
Genetic analysis: Sanger sequencing of the patient’s peripheral blood mononuclear cells performed in the early 90s detected no abnormalities in the IL2RG, ADA, and RAG1/2 genes, likely because of TME. Several years later, a more extensive panel of SCID-causing genes was sequenced using patient-derived EBV-transformed cell lines, a DNA source not impacted by TME. This time, a novel single base deletion in IL2RG causing a frameshift mutation was identified. However, because transformed lines are notorious for EBV-induced genetic aberrations this finding could not have been used as a definitive diagnosis. Sanger sequencing of maternal cells was normal for IL2RG, suggesting this might have been either a de-novo mutation or false-positive result. The next option for diagnosis was to obtain fibroblasts via a skin biopsy, or epithelial cells from a buccal smear.
Next generation sequencing (Stavropoulos et al. 2016) performed on a buccal smear showed that approximately 81% of the sequence contained the deletion in IL2RG and 19% of the sequence was wild-type. This result could be consistent with the fact that buccal-derived cells can be contaminated with engrafted donor cells, likely stemming from lymphocytes.
Sequencing performed in our patient’s sister did not detect the single base deletion in IL2RG. A similar analysis on the patient’s mother yielded 1 read out of 164 that showed the same deletion. This is an extremely low level of mosaicism and could be easily missed by performing traditional sequencing.
Discussion: Patients with SCID may have unknown genetic mutations explaining their phenotype. The importance of genetic analysis in family members of such patients needs to be emphasized for family planning and subsequent genetic counselling. Our patient underwent next generation sequencing (SCID panel) 2 decades following his SCID diagnosis, confirming a suspected mutation in IL2RG. Subsequently, his mother and sister underwent molecular genetic testing which revealed mosaicism in the mother and no deletion in the sister. As the sister had no deletion detected, her risk of being mosaic was extremely low and she is therefore unlikely to pass on the mutation to her children.
Our patient poses no risk to passing on his mutation to his male children given his X-linked condition, but his risk of passing on the mutation to his female children is 100%. Thus, none of the patient’s offspring will develop SCID, but female descendants will be carriers of the pathogenic variant.
This case report emphasizes the complexity of genetic analysis in SCID patients and their family members, and the importance of pursuing a molecular diagnosis. Next generation sequencing appears superior to traditional methods in providing answers for family planning and subsequent genetic counselling.
REFERENCES
Fischer, A., Cavazzana-Calvo, M., De Saint Basile, G., DeVillartay, J.P., Di Santo, J.P., Hivroz, C., Rieux-Laucat, F., and Le Deist, F. 1997. Naturally occurring primary deficiencies of the immune system. Annu. Rev. Immunol. 15:93–124. PMID: https://pubmed.ncbi.nlm.nih.gov/9143683/. doi: https://doi.org/10.1146/annurev.immunol.15.1.93.
Kwan, A., Abraham, R.S., Currier, R., Brower, A., Andruszewski, K., Abbott, J.K., Baker, M., Ballow, M., Bartoshesky, L.E., Bonilla, F.A., Brokopp, C., Brooks, E., Caggana, M., Celestin, J., Church, J.A., Comeau, A.M., Connelly, J.A., Cowan, M.J., Cunningham-Rundles, C., Dasu, T., Dave, N., De La Morena, M.T., Duffner, U., Fong, C.T., Forbes, L., Freedenberg, D., Gelfand, E.W., Hale, J.E., Hanson, I.C., Hay, B.N., Hu, D., Infante, A., Johnson, D., Kapoor, N., Kay, D.M., Kohn, D.B., Lee, R., Lehman, H., Lin, Z., Lorey, F., Abdel-Mageed, A., Manning, A., McGhee, S., Moore, T.B., Naides, S.J., Notarangelo, L.D., Orange, J.S., Pai, S.Y., Porteus, M., Rodriguez, R., Romberg, N., Routes, J., Ruehle, M., Rubenstein, A., Saavedra-Matiz, C.A., Scott, G., Scott, P.M., Secord, E., Seroogy, C., Shearer, W.T., Siegel, S., Silvers, S.K., Stiehm, E.R., Sugerman, R.W., Sullivan, J.L., Tanksley, S., Tierce, M.L. IV, Verbsky, J., Vogel, B., Walker, R., Walkovich, K., Walter, J.E., Wasserman, R.L., Watson, M.S., Weinberg, G.A., Weiner, L.B., Wood, H., Yates, A.B., Puck, J.M., and Bonagura, V.R. 2014. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 312:729–738. PMID: https://pubmed.ncbi.nlm.nih.gov/25138334/. doi: https://doi.org/10.1001/jama.2014.9132.
Liu, C., Duffy, B., Bednarski, J.J., Calhoun, C., Lay, L., Rundblad, B., Payton, J.E., and Mohanakumar, T. 2016. Maternal T-cell engraftment interferes with human leukocyte antigen typing in severe combined immunodeficiency. Am. J. Clin. Pathol. 145:251–257. PMID: https://pubmed.ncbi.nlm.nih.gov/26834123/. doi: https://doi.org/10.1093/ajcp/aqv079.
Shearer, W.T., Dunn, E., Notarangelo, L.D., Dvorak, C.C., Puck, J.M., Logan, B.R., Griffith, L.M., Kohn, D.B., O’Reilly, R.J., Fleisher, T.A., Pai, S.Y., Martinez, C.A., Buckley, R.H., and Cowan, M.J. 2014. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: The Primary Immune Deficiency Treatment Consortium experience. J. Allergy Clin. Immunol. 133:1092–1098. PMID: https://pubmed.ncbi.nlm.nih.gov/24290292/. doi: https://doi.org/10.1016/j.jaci.2013.09.044.
Stavropoulos, D.J., Merico, D., Jobling, R., Bowdin, S., Monfared, N., Thiruvahindrapuram, B., Nalpathamkalam, T., Pellecchia, G., Yuen, R.K.C., Szego, M.J., Hayeems, R.Z., Shaul, R.Z., Brudno, M., Girdea, M., Frey, B., Alipanahi, B., Ahmed, S., Babul-Hirji, R., Porras, R.B., Carter, M.T., Chad, L., Chaudhry, A., Chitayat, D., Doust, S.J., Cytrynbaum, C., Dupuis, L., Ejaz, R., Fishman, L., Guerin, A., Hashemi, B., Helal, M., Hewson, S., Inbar-Feigenberg, M., Kannu, P., Karp, N., Kim, R., Kronick, J., Liston, E., MacDonald, H., Mercimek-Mahmutoglu, S., Mendoza-Londono, R., Nasr, E., Nimmo, G., Parkinson, N., Quercia, N., Raiman, J., Roifman, M., Schulze, A., Shugar, A., Shuman, C., Sinajon, P., Siriwardena, K., Weksberg, R., Yoon, G., Carew, C., Erickson, R., Leach, R.A., Klein, R., Ray, P.N., Meyn, M.S., Scherer, S.W., Cohn, R.D., and Marshall, C.R. 2016. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genom. Med. 1:15012. PMID: https://pubmed.ncbi.nlm.nih.gov/28567303/. doi: https://doi.org/10.1038/npjgenmed.2015.12.
Wahlstrom, J.T., Dvorak, C.C., and Cowan, M.J. 2015. Hematopoietic stem cell transplantation for severe combined immunodeficiency. Curr. Pediatr. Rep. 3:1–10. PMID: https://pubmed.ncbi.nlm.nih.gov/25821657/. doi: https://doi.org/10.1007/s40124-014-0071-7.
Wahlstrom, J.T., Patel, K., Eckhert, E., Kong, D., Horn, B., Cowan, M.J., and Dvorak, C.C. 2017. Transplacental maternal engraftment and posttransplantation graft-versus-host disease in children with severe combined immunodeficiency. J. Allergy Clin. Immunol. 139:628–633.e10. PMID: https://pubmed.ncbi.nlm.nih.gov/27444177/. doi: https://doi.org/10.1016/j.jaci.2016.04.049.
Coronin 1A deficiency: first presentation as a positive newborn screen for severe combined immunodeficiency
Yael Dinur Schejtera,b, Adi Ovadiaa,b, Harjit Dadia,b, Chaim M. Roifmana,b
aDivision of Immunology and Allergy, Department of Paediatrics, The Hospital for Sick Children, Toronto, ON, Canada; bCanadian Centre for Primary Immunodeficiency, The Jeffrey Modell Research Laboratory for the Diagnosis of Primary Immunodeficiency, The Hospital for Sick Children and The University of Toronto, Toronto, ON, Canada
Background: Coronin 1A belongs to a large family of highly conserved actin regulatory proteins (Xavier et al. 2008). This protein has a clear role in T cell homeostasis both in mice and humans (Shiow et al. 2008), although the exact mechanism is yet to be clarified. A role for Coronin 1A was also observed in macrophages (Jayachandran et al. 2007), NK (Mace and Orange 2014), and neuronal cells (Martorella et al. 2017).
Human Coronin 1A deficiency was first described in a patient who presented with severe combined immunodeficiency (SCID). The patient received a hematopoietic stem cell transplant (HSCT) at the age of 4 years, with full lymphoid and myeloid donor chimerism (Shiow et al. 2008, 2009), but details of her long term follow up are not available. Later on, several reported cases extended the spectrum of disease associated with mutations in Coronin 1A (Moshous et al. 2013; Mace and Orange 2014; Stray-Pedersen et al. 2014): Hypomorphic mutations in 3 siblings were associated with a predisposition for EBV-mediated B cell lymphoproliferation at an early age (Moshous et al. 2013). The older sibling was reported to be in stable remission more than 10 years post-treatment of his EBV-associated lymphoma with no HSCT. The 2 younger siblings died during induction of chemotherapy and 4 months post-HSCT, respectively. An additional kindred with a mutation resulting in complete loss of protein expression (Stray-Pedersen et al. 2014), presented with a late onset disease at 7 years of age, with epidermodysplasia-verruciformis-human-papilloma-virus (EV-HPV), molluscum contagiosum and mucocutaneous herpetic ulcers, as well as granulomatous tuberculoid leprosy. The first patient developed EBV-associated lymphomas at 15 years of age and died despite aggressive treatment, including a haploidentical HSCT from her mother. Her younger brother is reported to be planned for HSCT. Abnormalities in NK cell cytotoxic function in one of these patients were identified (Mace and Orange 2014). Interestingly, both asymptomatic carrier parents were found to have immune abnormalities, including CD4+ and NK cell lymphopenia.
We report here a case of coronin 1A deficiency detected by newborn screening for SCID. To the best of our knowledge, this is the first reported case of Coronin 1A detected after birth by T cell receptor excision circle (TREC)-based newborn screening.
Methods: Patient information was collected prospectively and retrospectively from medical records.
Exome Sequencing and Variant Calling as well as Western blotting were done according to standard protocols. Sequencing analysis by polymerase chain reaction (PCR) with specific primers designed upstream and downstream of the Coronin 1A gene.
Case Presentation: The patient is a 14-month-old female, born at term to a single mother of African descent. Perinatal history was unremarkable. There is no known consanguinity in the family. The patient was found to have low TREC values in a newborn screening program. An initial TREC value was 42 copies/3 μL. A repeat test from the same dried blood sample was abnormal at 17 copies/3 μL (cutoff values >75 copies/3 μL). Screening for TBX deletion and purine profile was normal.
Immune evaluation: Since birth, the patient has had persistent lymphopenia as well as neutropenia. Immune work up further revealed a reduction in B and T cell counts, with a relatively more profound CD8+ cell lymphopenia. Over time, the patient developed a mild reduction in NK cell counts as well. Her T cell responses to mitogens were normal. A humoral work up showed hypogammaglobulinemia with good specific response to tetanus vaccine.
Whole exome sequencing (Stavropoulos et al. 2016, Wang et al. 2010) identified a novel homozygous mutation in Coronin 1A. The mother was found to be a heterozygous carrier.
Discussion: We report here the first Coronin 1A deficiency patient detected by newborn screening for SCID. This patient had leukopenia and neutropenia, but is currently doing clinically well at 14 months of age with no severe or recurrent infections. Previous reports show great variability in clinical presentations, ranging from patients presenting as SCID (Shiow et al. 2008), through EBV-related lymphoproliferation at a young age (Moshous et al. 2013), and a yet later onset of disease, at 7 years of age (Stray-Pedersen et al. 2014). All patients reported thus far presented with T cell lymphopenia (Shiow et al. 2008; Moshous et al. 2013; Stray-Pedersen et al. 2014) and a severe reduction in CD45Ra+ naïve T cells (Moshous et al. 2013; Moshous and de Villartay 2014; Stray-Pedersen et al. 2014), suggesting a role for Coronin 1A in mature T cell survival. However, B and NK cell counts, T cell responses to mitogens and antigens, as well as humoral function are all variable among patients (Shiow et al. 2008; Moshous et al. 2013; Mace and Orange 2014; Stray-Pedersen et al. 2014). One patient (Mace and Orange 2014) was also found to have abnormalities in NK cytotoxic function.
Summary: We hereby report the first case of Coronin 1A deficiency presenting in a well newborn as part of the newborn screening program. Coronin 1A deficiency is a rare combined immunodeficiency, and the few cases reported in the literature had a variable although detrimental clinical course. As our patient is currently well, we are confronted with one of the challenges posed by early diagnosis of rare diseases, i.e., the inability of predicting prognosis, and thus the difficulty in recommending a morbidity and mortality associated treatment, such as HSCT.
REFERENCES
Jayachandran, R., Sundaramurthy, V., Combaluzier, B., Mueller, P., Korf, H., Huygen, K., Miyazaki, T., Albrecht, I., Massner, J., and Pieters, J. 2007. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 130(1):37–50. PMID: https://pubmed.ncbi.nlm.nih.gov/17632055/. doi: https://doi.org/10.1016/j.cell.2007.04.043.
Mace, E.M., and Orange, J.S. 2014. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proc. Natl. Acad. Sci. U.S.A. 111(18):6708–6713. PMID: https://pubmed.ncbi.nlm.nih.gov/24760828/. doi: https://doi.org/10.1073/pnas.1314975111.
Martorella, M., Barford, K., Winkler, B., and Deppmann, C.D. 2017. Emergent role of Coronin-1a in neuronal signaling. Vitam. Horm. 104:113–131. PMID: https://pubmed.ncbi.nlm.nih.gov/28215292/. doi: https://doi.org/10.1016/bs.vh.2016.10.002.
Moshous, D., and de Villartay, J.-P. 2014. The expanding spectrum of human coronin 1A deficiency. Curr. Allergy Asthma Rep. 14(12):481. PMID: https://pubmed.ncbi.nlm.nih.gov/25269405/. doi: https://doi.org/10.1007/s11882-014-0481-1.
Moshous, D., Martin, E., Carpentier, W., Lim, A., Callebaut, I., Canioni, D., Hauck, F., Majewski, J., Schwartzentruber, J., Nitschke, P., Sirvent, N., Frange, P., Picard, C., Blanche, S., Revy, P., Fischer, A., Latour, S., Jabado, N., and de Villartay, J.P. 2013. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J. Allergy Clin. Immunol. 131(6):1594–1603. PMID: https://pubmed.ncbi.nlm.nih.gov/23522482/. doi: https://doi.org/10.1016/j.jaci.2013.01.042.
Shiow, L.R., Paris, K., Akana, M.C., Cyster, J.G., Sorensen, R.U., and Puck, J.M. 2009. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a coronin-1A mutation and a chromosome 16p11.2 deletion. Clin. Immunol. 131(1):24–30. PMID: https://pubmed.ncbi.nlm.nih.gov/19097825/. doi: https://doi.org/10.1016/j.clim.2008.11.002.
Shiow, L.R., Roadcap, D.W., Paris, K., Watson, S.R., Grigorova, I.L., Lebet, T., An, J., Xu, Y., Jenne, C.N., Föger, N., Sorensen, R.U., Goodnow, C.C., Bear, J.E., Puck, J.M., and Cyster, J.G. 2008. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat. Immunol. 9(11):1307–1315. PMID: https://pubmed.ncbi.nlm.nih.gov/18836449/. doi: https://doi.org/10.1038/ni.1662.
Stavropoulos, D.J., Merico, D., Jobling, R., Bowdin, S., Monfared, N., Thiruvahindrapuram, B., Nalpathamkalam, T., Pellecchia, G., Yuen, R.K.C., Szego, M.J., Hayeems, R.Z., Shaul, R.Z., Brudno, M., Girdea, M., Frey, B., Alipanahi, B., Ahmed, S., Babul-Hirji, R., Porras, R.B., Carter, M.T., Chad, L., Chaudhry, A., Chitayat, D., Doust, S.J., Cytrynbaum, C., Dupuis, L., Ejaz, R., Fishman, L., Guerin, A., Hashemi, B., Helal, M., Hewson, S., Inbar-Feigenberg, M., Kannu, P., Karp, N., Kim, R., Kronick, J., Liston, E., MacDonald, H., Mercimek-Mahmutoglu, S., Mendoza-Londono, R., Nasr, E., Nimmo, G., Parkinson, N., Quercia, N., Raiman, J., Roifman, M., Schulze, A., Shugar, A., Shuman, C., Sinajon, P., Siriwardena, K., Weksberg, R., Yoon, G., Carew, C., Erickson, R., Leach, R.A., Klein, R., Ray, P.N., Meyn, M.S., Scherer, S.W., Cohn, R.D., and Marshall, C.R. 2016. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. NPJ Genom. Med. 1(1):15012. PMID: https://pubmed.ncbi.nlm.nih.gov/28567303/. doi: https://doi.org/10.1038/npjgenmed.2015.12.
Stray-Pedersen, A., Jouanguy, E., Crequer, A., Bertuch, A.A., Brown, B.S., Jhangiani, S.N., Muzny, D.M., Gambin, T., Sorte, H., Sasa, G., Metry, D., Campbell, J., Sockrider, M.M., Dishop, M.K., Scollard, D.M., Gibbs, R.A., Mace, E.M., Orange, J.S., Lupski, J.R., Casanova, J.L., and Noroski, L.M. 2014. Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. J. Clin. Immunol. 34(7):871–890. PMID: https://pubmed.ncbi.nlm.nih.gov/25073507/. doi: https://doi.org/10.1007/s10875-014-0074-8.
Wang, K., Li, M., and Hakonarson, H. 2010. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38(16):e164. PMID: https://pubmed.ncbi.nlm.nih.gov/20601685/. doi: https://doi.org/10.1093/nar/gkq603.
Xavier, C.-P., Eichinger, L., Fernandez, M.P., Morgan, R.O., and Clemen, C.S. 2008. Evolutionary and functional diversity of coronin proteins. Subcell. Biochem. 48:98–109. PMID: https://pubmed.ncbi.nlm.nih.gov/18925374/. doi: https://doi.org/10.1007/978-0-387-09595-0_9.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 4 • Number 4 • December 2017
Pages: 137 - 149
History
Version of record online: 21 December 2017
Copyright
© 2017.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
2017. Abstracts from the Immunodeficiency Canada—5th SCID Symposium, Toronto, ON, 12 October 2017. LymphoSign Journal.
4(4): 137-149. https://doi.org/10.14785/lymphosign-2017-0013
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
