A mutation in the STAT1 DNA-binding domain associated with hemophagocytic lymphohistocytosis
Abstract
Introduction: The transcription factor Signal Transducer and Activator of Transcription 1 (STAT1) is a key element in many of the signalling cascades involved in immune system function. Different mutations in STAT1 are associated with heterogeneous clinical phenotypes that range from early fatality due to overwhelming infection to limited involvement of the mucus membrane with recurrent Candida infections. Multiple genes related to immune function have been associated with the development of hemophagocytic lymphohistiocytosis (HLH), but the association between STAT1 mutation and HLH has not been described in detail.
Methods: We report the genetic background of a patient with chronic mucocutaneous candidiasis (CMC) as well as an unusual clinical course.
Results: In this study we describe a patient with a mutation in the STAT1 DNA-binding domain and a history of CMC who developed a refractory and fatal case of HLH despite having bone marrow transplantation.
Conclusion: We describe a patient with refractory and fatal HLH who was found to have a mutation in the DNA-binding domain of STAT1.
Statement of novelty: The association of chronic mucocutaneous candidiasis with HLH.
Introduction
The DNA-binding protein Signal Transducer and Activator of Transcription 1 (STAT1) is a member of a family of transcription factors that plays a major role in different and diverse functions of the cell. Following activation by interferon-gamma or interferon-alpha, STAT1 protein is phosphorylated and forms dimers. It then translocates into the nucleus, binds to DNA, and initiates its effector function (Darnell et al. 1994; Stark et al. 1998; Levy and Darnell 2002). Different genetic mutations in STAT1 resulting in different clinical phenotypes have been described (Dupuis et al. 2001, 2003; Chapgier et al. 2006a, 2006b). Patients with complete loss-of-function of the STAT1 protein usually die in childhood from overwhelming viral and mycobacterial infections (Dupuis et al. 2003). Other patients, who have the autosomal dominant STAT1 loss-of-function form, usually present with a predisposition to low pathogenic mycobacterial infections (Tsumura et al. 2012). Different mutations, mostly in the coiled–coiled domain, are believed to lead to gain of function of the STAT1 protein and cause an autosomal dominant form of chronic mucocutaneous candidiasis (CMC) (Liu et al. 2011; van de Veerdonk et al. 2011). Recently, a severe and fatal form of combined immunodeficiency was found to be associated with a declining immunity during childhood, caused by predominantly de-novo mutations in the STAT1 DNA-binding domain (DBD) (Sharfe et al. 2014).
Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening inflammatory process that is characterized by uncontrolled activation of macrophages and histiocytes (Favara et al. 1997). Traditionally, HLH was divided into a genetic form (also known as the familial or primary type) and an acquired form (secondary HLH) (Writing Group of the Histiocyte Society 1987; Henter et al. 1991).
The genetic form usually presents in a young infant with parental consanguinity or a history of other affected family members. Patients in this group have a genetic defect in genes that are responsible for the cytotoxic function of T cells and NK cells. Most commonly, mutations in the gene PFR1 that encodes for Perforin1, are detected in this group of patients; however, other genes, such as UNC13D and STX11 that encode for cytolytic granule trafficking and exocytosis may also be associated with the aberrant inflammation (Jordan et al. 2011). Although some of the primary immunodeficiency disorders that are associated with HLH have abnormal cytotoxic function of T cells and NK cells (for example Chédiak–Higashi syndrome and Griscelli syndrome type 2) others such as Wiskott–Aldrich and DiGeorge syndrome do not, and the mechanism leading to HLH in these conditions is not fully understood (Introne et al. 1999; Ménasché et al. 2000; Cesaro et al. 2003; Pasic et al. 2003).
In contrast to the genetic form of HLH, the acquired forms seem not to be associated with a known genetic abnormality or immunodeficiency syndrome. As in some of the genetic forms, the pathogenesis is not completely understood, and it seems that the disease appears in association with infection, malignancy, rheumatic conditions, metabolic disorders, or as a consequence of drug toxicity (Henter et al. 1991; Janka et al. 1998). Nevertheless, it is conceivable that modifier genes may predispose individuals to developing secondary HLH. The symptoms and signs of HLH are nonspecific; fever, hepatosplenomegaly, lymphadenopathy, cytopenia, morbiliformic rash, and in some cases involvement of the central nervous system (CNS) with seizures or encephalopathy (Henter et al. 2007).
Without treatment, the prognosis of HLH is poor, with less than 5% survival 1 year post diagnosis (Janka 1983). Even with appropriate treatment, the mortality rate can be as high as 45% (Henter et al. 2002). The main goal of the treatment is to suppress the exaggerated activation of the immune system with immunosuppressive agents such as dexamethasone, etoposide, and cyclosporine, and when needed and indicated a bone marrow transplantation (Henter et al. 2002).
We report here an occurrence of fatal HLH in a patient with CMC due to STAT1 deficiency.
Methods
Flow cytometry
Peripheral blood mononuclear cells were obtained by Ficoll–Hypaque density gradient centrifugation, and surface phenotypes were determined by flow cytometry on a Coulter EPICS V flow cytometer (Beckman Coulter, Brea, Calif.), with a single argon laser, which analyzes up to 3 colours simultaneously. Single colour and (or) isotype antibody controls were both used for multicolour staining.
Proliferation assay
Lymphocyte proliferative responses to mitogens (including phytohemagglutinin and anti-CD3 antibodies) and to a panel of recall antigens (including Candida, tetanus, Herpes zoster, and cytomegalovirus) were determined by thymidine incorporation. All assays were performed in triplicate and were compared with simultaneously stimulated random normal controls.
DNA sequencing
Genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by polymerase chain reaction (PCR) with specific primers for exons 1–23 of the coding sequence of STAT1 (NM_007315.3) (available upon request). Exons and their flanking intronic regions were sequenced with the GenomeLab Dye Terminator Cycle Sequencing Quick Start Kit (Beckman Coulter) and analyzed on a CEQ 8000 Genetic Analysis System (Beckman Coulter).
Results
Case presentation
The patient, a female of nonconsanguineous East Indian origin, presented at 5 months of age with recurrent episodes of skin infection and persistent diaper rash and oral thrush. Her past medical history was unremarkable and her family history was negative for immunodeficiency, malignancy, or autoimmune disorders. The laboratory findings were unremarkable with normal levels of leukocytes and immunoglobulins (Table 1). She responded to Nystatin but continued to have periodic episodes of oral thrush. At the age of 3 years she was diagnosed with hypothyroidism and began suffering repeated episodes of pneumonia as well as asthma. At the age of 9 years she was found to have oral thrush and herpetic vesicles on her chest without hepatosplenomegaly or lymphadenopathy. She responded well to treatment with oral Fluconazole and Acyclovir. She subsequently developed Coombs-positive hemolytic anemia.
Table 1:
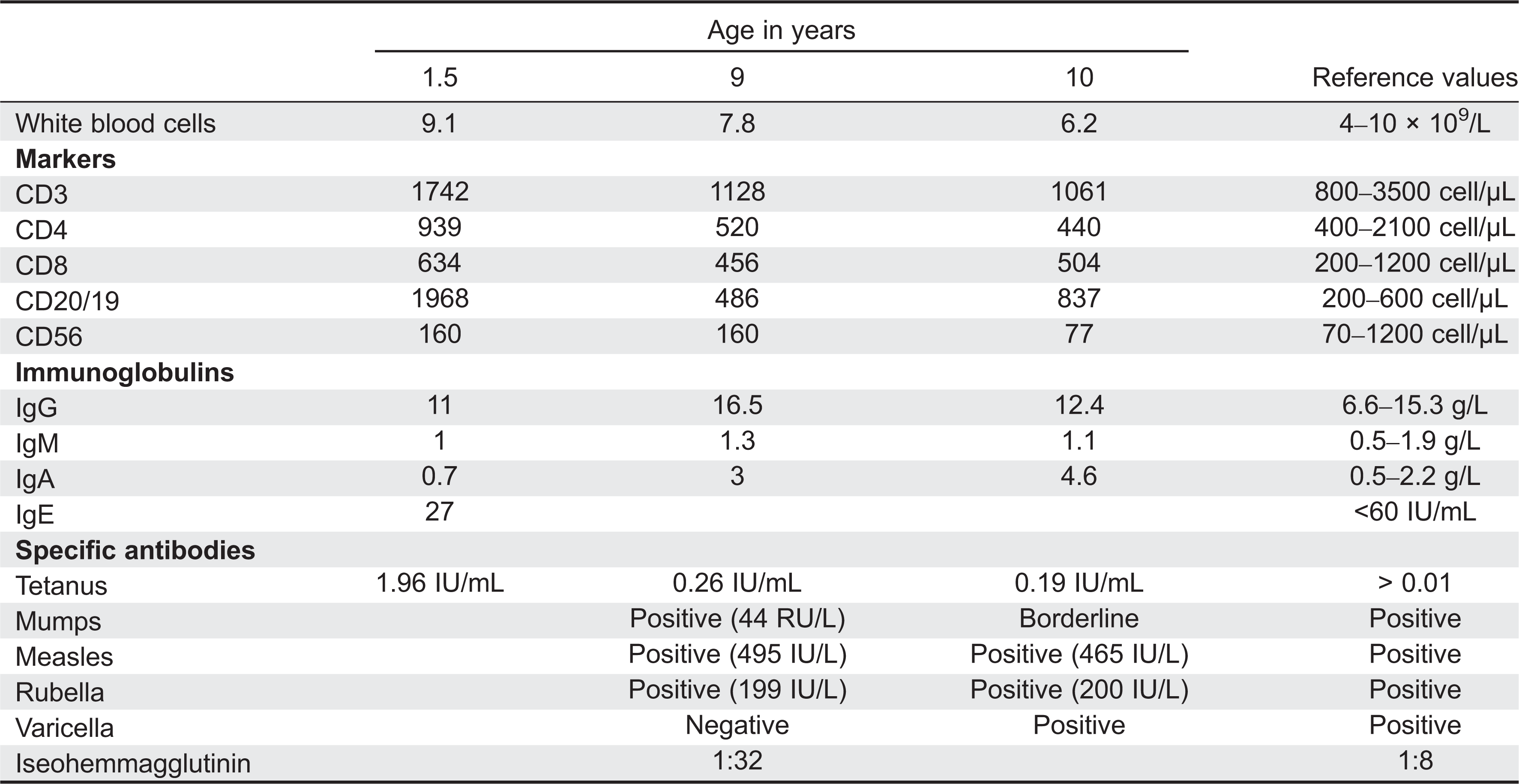
Evaluation of the immune system
Evaluation of the immune system revealed normal numbers of circulating white blood cells and lymphocytes. Flow cytometry analysis showed a steady decline of CD3+ circulating lymphocytes over time. Predominantly CD4+ T cells diminished over time down to 440 cells/µL. In addition, the number of NK cells was also declining to 77 cells/µL at the age of 10 years. These changes are consistent with the recently documented decline in immunity in patients with STAT1 mutations in the DBD (Sharfe et al. 2014).
The number of circulating B cells appears to have been normal with little decline. Serum immunoglobulin levels increased over time particularly for IgG and IgA (Table 1), and specific antibodies to vaccination appeared to be preserved. In-vitro response to mitogens and antigens were still preserved in this patient.
Genetic analysis
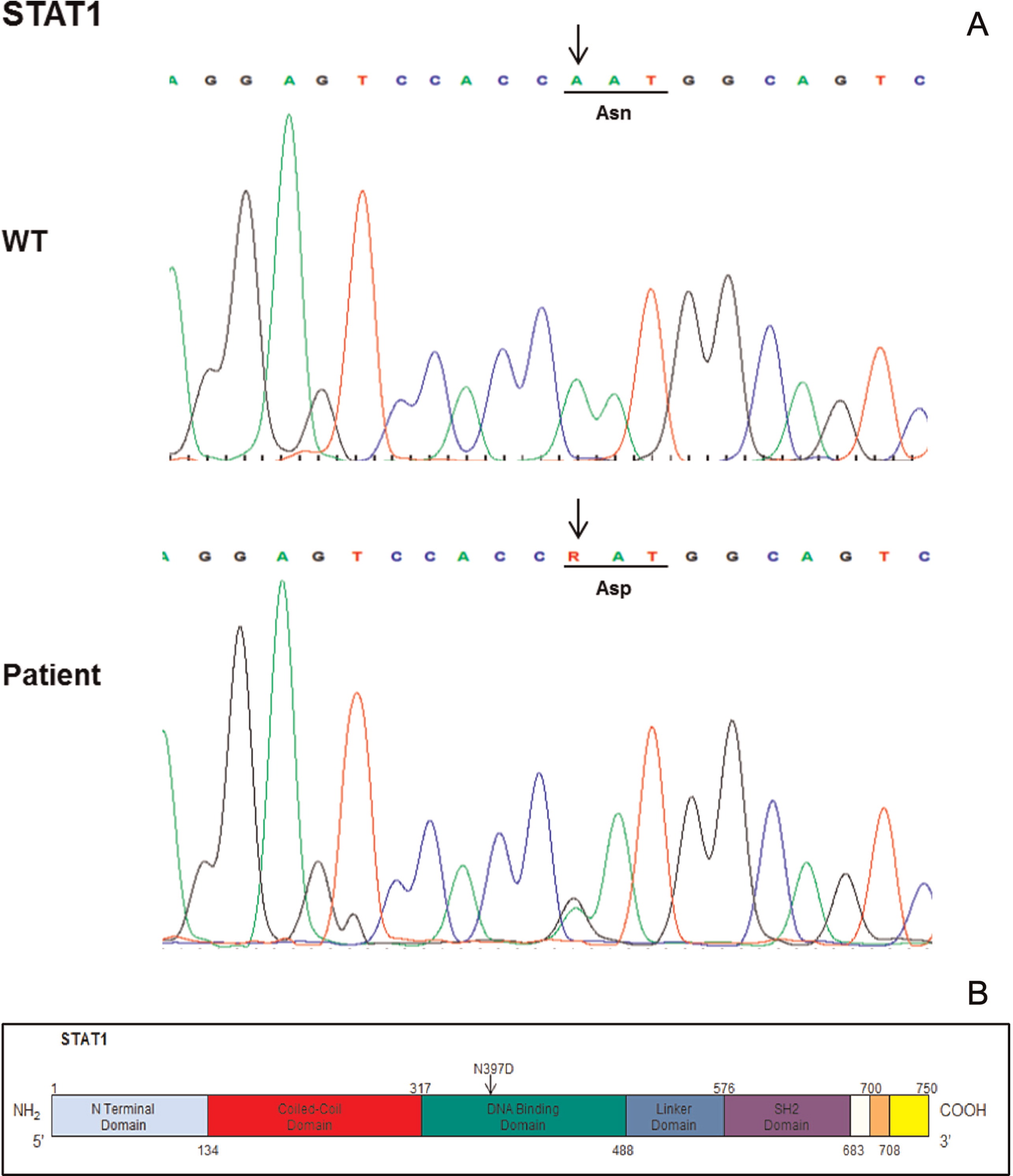
Genetic analysis of the STAT1 gene (Figure 1) revealed a mutation in the DBD of the molecule. The mutation was a de novo mutation, as it was not present in the parents or in the brother of the patient.
Figure 1:
Course of HLH
The patient presented at the age of 10 years in the emergency department with a high fever, cough, and pancytopenia. An abdominal ultrasound and abdominal CT (taken later), showed findings suggestive of a micro-abscess in the kidneys and spleen. An extensive infection workup that included blood cultures, serology, and PCR testing for bacteria, mycobacteria, fungi, and different viruses (Herpes family, respiratory viruses, and parvovirus) identified Epstein–Barr Virus viremia with a viral load of 1127 copies/mL in the blood (but not in the plasma). In addition, Herpes zoster virus was isolated from a skin vesicle.
As the fever continued, an initial blood work was sent to assess for HLH. The diagnostic criteria for HLH, according to the HLH-2004 protocol by the Histiocyte Society, include a molecular diagnosis consistent with HLH or 5 out of 8 clinical and laboratory criteria as described in Table 2 (Henter et al. 2007).
Table 2:

Although the initial blood work didn't fully support the diagnosis of HLH, the repeated blood work that was done on the second week of her admission confirmed the diagnosis (Table 3).
Table 3:
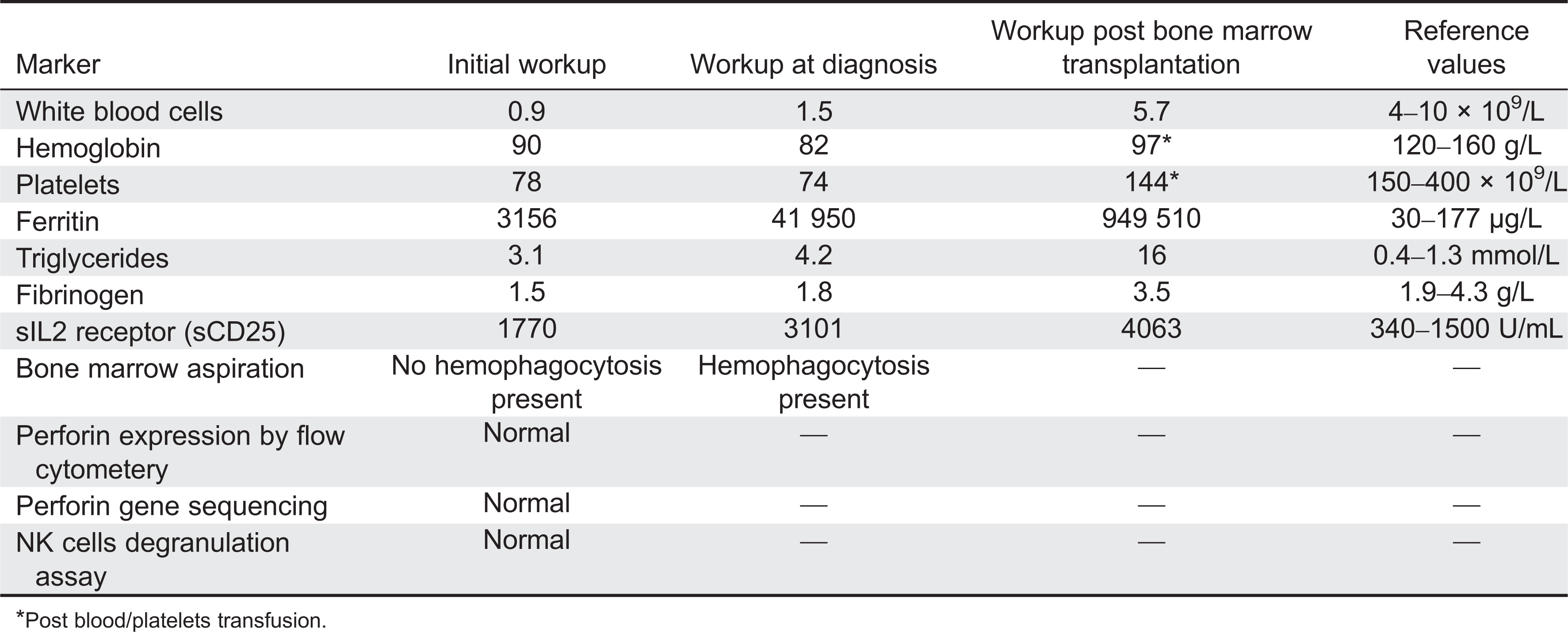
*
Post blood/platelets transfusion.
The patient was treated with broad spectrum antibiotics, anti-fungal, and anti-viral medications. After HLH was diagnosed, she started treatment with dexamethasone but showed only partial response. A day following initial steroid treatment she developed seizures and her brain MRI showed bilateral multifocal abnormal signals in the cortex and subcortical white matter associated with leptomeningeal enhancement, findings that are consistent but not specific to CNS involvement of HLH (Figure 2).
Figure 2:
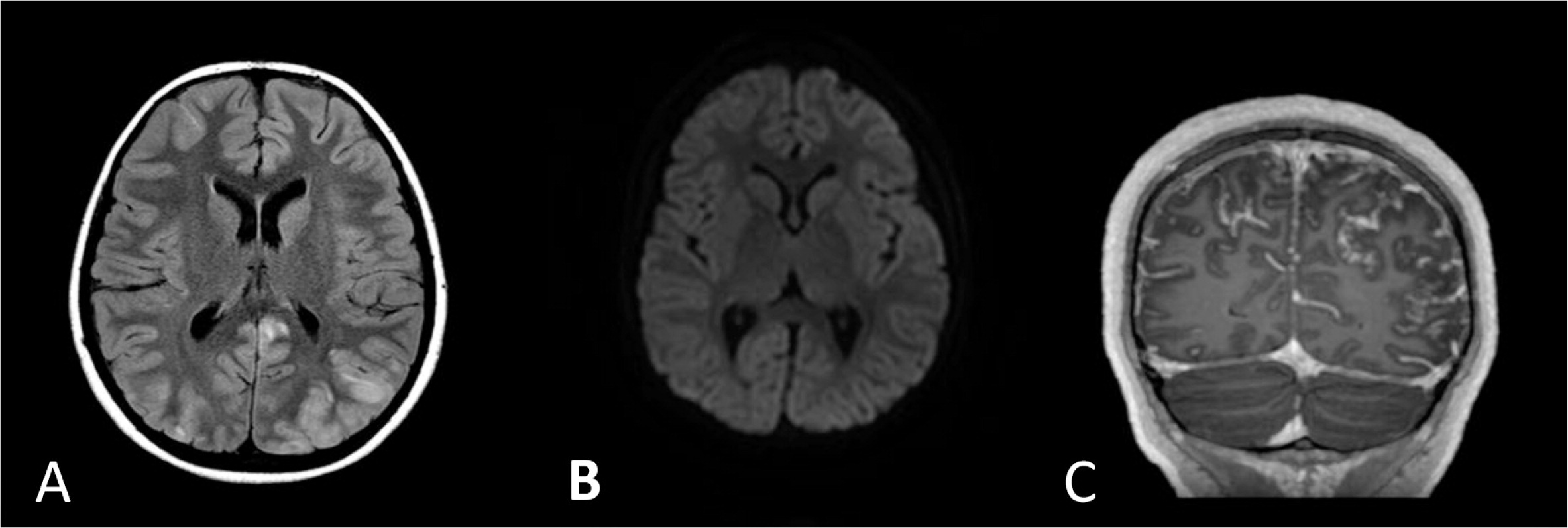
Despite receiving appropriate antibiotic treatment, she did not improve and continued to be febrile with increased laboratory markers of HLH. The treatment for HLH was escalated to include etoposide, cyclosporine, and intrathecal methotrexate similar to the HLH-2004 protocol recommendation. The patient had a temporary response to the treatment as fevers subsided and repeated head imaging showed resolution of the abnormal MRI findings (Figures 3 and 4). However, she failed to get into full remission as her laboratory markers persistently showed cytopenia, increased ferritin, and elevated sCD25 levels.
Figure 3:
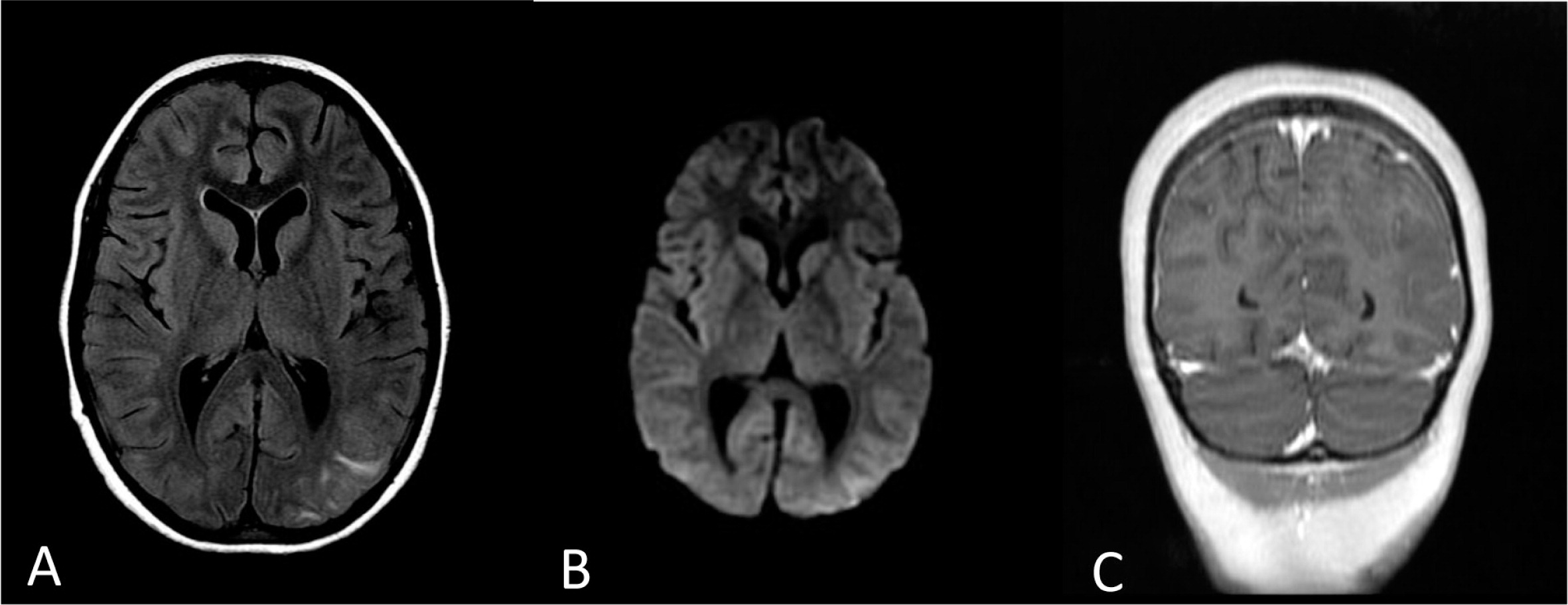
Figure 4:
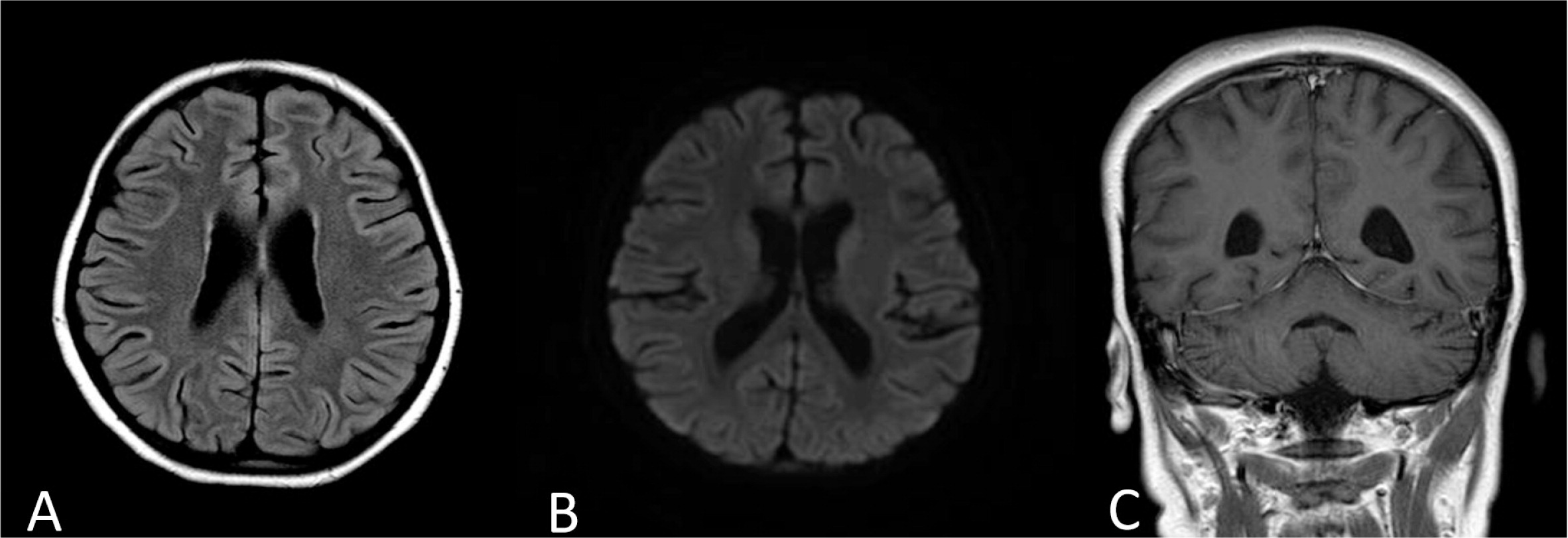
After 8 weeks of treatment and following the HLH-2004 protocol recommendation for severe cases of HLH, a bone marrow transplant (BMT) was performed using an HLA matched unrelated donor in the absence of a matched sibling. The patient received a full myeloablative conditioning that included Busulfan (3.6 mg/kg) on days −8 to −5, etoposide (30 mg/kg/dose) on day −4, and cyclophosphamide (60 mg/kg) on days −3 and −2. For graft versus host disease prophylaxis she received steroids, cyclosporine, and methotrexate. The patient fully engrafted on day +8 with 100% chimerism, but she spiked a fever and once again suffered seizures within a week of transplant. A repeat MRI of her head again showed multiple cortical and white matter lesions, similar to the lesions described in Figure 2. The laboratory markers for HLH, which are sCD25, sCD163, and ferritin peaked to 4063 U/mL, 8178 ng/mL, and 949 510 µg/L, respectively. She subsequently developed severe complications including gastrointestinal bleeding, pulmonary hemorrhage, toxic epidermal necrosis, and renal failure. In addition, alemtuzumab was added to the regimen with no response. The patient deteriorated gradually until her death from multi-organ failure.
Discussion
In the last few years, different mutations in the STAT1 gene have been reported (Liu et al. 2011; van de Veerdonk et al. 2011; Tsumura et al. 2012). Monoallelic mutations in the DBD of STAT1 seem to cause a severe and frequently fatal immunodeficiency (Sharfe et al. 2014). In this study we described a mutation in the DBD of the molecule. The fact that these mutations are de novo mutations, support the hypothesis that changes in the DBD of the STAT1 gene are lethal at a young age before the carrier has a chance to produce offspring. The patient we described here had clinical symptoms of CMC and declining immunity that specifically displayed a decline in CD4+ T cells and NK cells. The diminished number and function of NK cells were previously reported to be associated with HLH. In this case, it is also conceivable that the decline in NK cell numbers underscored at least in part the development of HLH, although the NK cell degranulation assay was normal.
CNS involvement is common in HLH patients, and symptoms can range from abnormal findings in the cerebrospinal fluid to altered mental status, seizures, and coma. Active HLH, infection, and posterior reversible encephalopathy syndrome (PRES) are in the differential diagnosis in HLH patients with CNS involvement (Haddad et al. 1997; Goo and Weon 2007; Horne et al. 2008; Lee et al. 2013).
The overall survival of children with HLH who were treated with the HLH-94 protocol is around 55% and two-thirds of those who survived underwent BMT. It appears that survival after BMT in HLH cases is fair, regardless of whether a HLA matched sibling or an unrelated match donor are used (Henter et al. 2007). Although there are some reports that support reduced intensity over full myeloablative conditioning (Cooper et al. 2006; Marsh et al. 2010), at present there is insufficient evidence to support the use of one protocol over another. Out of the few reports of BMT in patients with STAT1 mutation, only 2 patients have been reported with long-term survival after receiving a BMT from a match sibling donor (Deeg et al. 1986; Hoh et al. 1996), whereas 2 other patients died after the procedure (Aldave et al. 2013).
In conclusion, we have shown an association between a monoallelic STAT1 mutation and a severe and refractory case of HLH. A thorough immunologic workup that includes STAT1 genetic analysis is needed in severe and refractory cases of HLH to identify those who have this uncommon genetic abnormality. More studies are in order to define optimal treatment options for patients with STAT1 mutation and HLH.
REFERENCES
Aldave J.C., Cachay E., Núñez L., Chunga A., Murillo S., Cypowyj S., et al. A 1-year-old girl with a gain-of-function STAT1 mutation treated with hematopoietic stem cell transplantation J. Clin. Immuno. 2013 33 8 1273 -1275
Cesaro S., Messina C., Sainati L., Danesino C., and Aricò M. Del 22Q11.2 and hemophagocytic lymphohistiocytosis: a non-random association Am. J. Med. Genet. . 2003 116A 2 208 -209
Chapgier A., Boisson-Dupuis S., Jouanguy E., Vogt G., Feinberg J., Prochnicka-Chalufour A., et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease PLoS Gene. 2006a 2 8 e131
Chapgier A., Wynn R.F., Jouanguy E., Filipe-Santos O., Zhang S., Feinberg J., et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo J. Immuno. 2006b 176 8 5078 -5083
Cooper N., Rao K., Gilmour K., Hadad L., Adams S., Cale C., et al. Stem cell transplantation with reduced-intensity conditioning for hemophagocytic lymphohistiocytosis Bloo. 2006 107 3 1233 -1236
Darnell J.E. Jr, Kerr I.M., and Stark G.R. Jak TAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins Scienc. 1994 264 5164 1415 -1421
Deeg H.J., Lum L.G., Sanders J., Levy G.J., Sullivan K.M., Beatty P., et al. Severe aplastic anemia associated with chronic mucocutaneous candidiasis. Immunologicand hematologicreconstitution after allogeneic bone marrow transplantation Transplantatio. 1986 41 5 583 -586
Dupuis S., Dargemont C., Fieschi C., Thomassin N., Rosenzweig S., Harris J., et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation Scienc. 2001 293 5528 300 -303
Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I.Z., Al-Jumaah S., et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Gene. 2003 33 3 388 -391
Favara B.E., Feller A.C., Pauli M., Jaffe E.S., Weiss L.M., Arico M., et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society Med. Pediatr. Onco. 1997 29 3 157 -166
Goo H.W. and Weon Y.C. A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis Pediatr. Radio. 2007 37 11 1110 -1117
Haddad E., Sulis M.L., Jabado N., Blanche S., Fischer A., and Tardieu M. Frequency and severity of central nervous system lesions in hemophagocytic lymphohistiocytosis Bloo. 1997 89 3 794 -800
Henter J.I., Elinder G., and Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society Semin. Onco. 1991 18 1 29 -33
Henter J.I., Horne A., Aricó M., Egeler R.M., Filipovich A.H., Imashuku S., et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatr. Blood Cance. 2007 48 2 124 -1231
Henter J.I., Samuelsson-Horne A., Aricò M., Egeler R.M., Elinder G., Filipovich A.H., et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation Bloo. 2002 100 7 2367 -2373
Hoh M.C., Lin H.P., Chan L.L., and Lam S.K. Successful allogeneic bone marrow transplantation in severe chronic mucocutaneous candidiasis syndrome Bone Marrow Transplan. 1996 18 4 797 -800
Horne A., Trottestam H., Aricò M., Egeler R.M., Filipovich A.H., Gadner H., et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis Br. J. Haemato. 2008 140 3 327 -335
Introne W., Boissy R.E., and Gahl W.A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome Mol. Genet. Meta. 1999 68 2 283 -303
Janka G.E. Familial hemophagocytic lymphohistiocytosis Eur. J. Pediat. 1983 140 3 221 -230
Janka G., Imashuku S., Elinder G., Schneider M., and Henter J.I. Infection- and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol Oncol. Clin. North A. 1998 12 2 435 -444
Jordan M.B., Allen C.E., Weitzman S., Filipovich A.H., and McClain K.L. How I treat hemophagocytic lymphohistiocytosis Bloo. 2011 118 15 4041 -4052
Lee G., Lee S.E., Ryu K.H., Yoo E.S., et al. Posterior reversible encephalopathy syndrome in pediatric patients undergoing treatment for hemophagocytic lymphohistiocytosis: clinical outcomes and putative risk factors Blood Re. 2013 48 4 258 -265
Levy D.E. and Darnell J.E. Jr. Stats: transcriptional control and biological impact Nat. Rev. Mol. Cell. Bio. 2002 3 9 651 -662
Liu L., Okada S., Kong X.F., Kreins A.Y., Cypowyj S., Abhyankar A., et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis J. Exp. Me. 2011 208 8 1635 -1648
Marsh R.A., Vaughn G., Kim M.O., Li D., Jodele S., Joshi S., et al. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic lymphohistiocytosis undergoing allogeneic hematopoietic cell transplantation Bloo. 2010 116 26 5824 -5831
Ménasché G., Pastural E., Feldmann J., Certain S., Ersoy F., Dupuis S., et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Gene. 2000 25 2 173 -176
Pasic S., Micic D., and Kuzmanovic M. Epstein-Barr virus-associated haemophagocytic lymphohistiocytosis in Wiskott-Aldrich syndrome Acta. Paediat. 2003 92 7 859 -861
Sharfe N., Nahum A., Newell A., Dadi H., Ngan B., Pereira S.L., et al. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1 J. Allergy. Clin. Immuno. 2014 133 3 807 -817
Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., and Schreiber R.D. How cells respond to interferons. Annu. Rev Bioche. 1998 67 227 -264
Tsumura M., Okada S., Sakai H., Yasunaga S., Ohtsubo M., Murata T., et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with Mendelian susceptibility to mycobacterial disease Hum. Muta. 2012 33 9 1377 -1387
van de Veerdonk F.L., Plantinga T.S., Hoischen A., Smeekens S.P., Joosten L.A., Gilissen C., et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis N. Engl. J. Me. 2011 365 1 54 -61
Writing Group of the Histiocyte Society Histiocytosis syndromes in children Lance. 1987 1 8526 208 -209
Information & Authors
Information
Published In

LymphoSign Journal
Volume 1 • Number 2 • December 2014
Pages: 87 - 95
History
Received: 28 May 2014
Accepted: 29 May 2014
Accepted manuscript online: 24 June 2014
Version of record online: 24 June 2014
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
FaitelsonYoram, BatesAndrea, ShroffManohar, GrunebaumEyal, RoifmanChaim M., and NaqviAhmed. 2014. A mutation in the STAT1 DNA-binding domain associated with hemophagocytic lymphohistocytosis. LymphoSign Journal.
1(2): 87-95. https://doi.org/10.14785/lpsn-2014-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
Cited by
1. The effects of STAT1 dysfunction on the gut
View Options
View options
Login options
Check if you access through your login credentials or your institution to get full access on this article.
