Identification of novel compound heterozygous LRBA mutations associated with recurrent hemophagocytic lymphohistiocytosis and CNS manifestations
Abstract
Background: Lipopolysaccharide-responsive beige-like anchor (LRBA) is an intracellular protein that regulates the recycling of cytotoxic T lymphocyte-associated protein 4 (CTLA4), an immune checkpoint molecule which prevents ongoing activation of T cells. Deficiency of LRBA results in increased trafficking and degradation of CTLA4, and consequently, uncontrolled T cell responses. The phenotypic spectrum of LRBA deficiency arising from biallelic loss-of-function typically includes recurrent infections, autoimmunity, lymphoproliferation, chronic diarrhea, hypogammaglobulinemia, and cytopenia.
Aim: To report an atypical presentation of LRBA deficiency arising from a set of compound heterozygous LRBA variants, encompassing recurrent hemophagocytic lymphocytosis (HLH) and neurological manifestations.
Methods: Clinical data was gathered through retrospective chart review. Expanded genetic analysis including whole exome sequencing was performed.
Results: Our patient initially presented at age 15 months with fever, seizures, and encephalopathy. HLH-work-up showed bicytopenia, elevated ferritin and triglyceride, and low fibrinogen, however, he did not yet meet the diagnostic criteria for HLH. MRI brain and EEG at diagnosis was suggestive of acute necrotizing encephalopathy of childhood. He responded to pulsed IV methylprednisolone treatment with minimal residual neurological deficit on follow-up. At 36 months of age, he had a repeat presentation and rapidly deteriorated. He developed severe encephalopathy with fixed dilated pupils. Whole exome sequencing revealed a set of compound heterozygous missense variants in the LRBA gene, a novel c.2206A>T (p.R736W) and c.5989C>T (p.R1997C) variant.
Conclusion: Compound heterozygous mutations in the LRBA gene caused an atypical presentation of recurrent HLH with central nervous system (CNS) manifestations in our patient.
Statement of Novelty: We herein report a novel set of compound heterozygous mutations in LRBA with atypical presentation of recurrent HLH with CNS manifestations, thus expanding the known phenotypic spectrum of LRBA deficiency.
Introduction
Lipopolysaccharide-responsive beige-like anchor (LRBA) is an intracellular protein that prevents the trafficking and degradation of cytotoxic T lymphocyte-associated protein 4 (CTLA4), an essential immune checkpoint molecule which dampens T cell-mediated responses and regulates immune self-tolerance. CTLA4 impairs the classic CD28 signaling pathway by binding the costimulatory ligands CD80 and CD86, located on antigen presenting cells, leading to ligand internalization and degradation. Given the linked biology of LRBA and CTLA4, absence of either molecules result in uncontrolled T cell activation, differentiation, and effector responses (Lo et al. 2015).
LRBA deficiency was first reported in 2012 in 4 consanguineous families with humoral immunodeficiency and features of autoimmunity (Lopez-Herrera et al. 2012). The clinical presentation of LRBA deficiency has since expanded from the early descriptions of recurrent infections, autoimmunity, lymphoproliferation, chronic diarrhea, hypogammaglobulinemia and cytopenia, to include common variable immunodeficiency (CVID) and autoimmune lymphoproliferative syndrome (ALPS)-like phenotypes (Cagdas et al. 2019) (Kostel Bal et al. 2017). Patients with ALPS-like phenotype presented with symptoms of organomegaly, lymphoproliferation, and elevated double negative T cells.
To date, there have been scarce reports of atypical presentations of LRBA deficiency, such as hemophagocytic lymphohistiocytosis (HLH) (Ren et al. 2021) and neurological manifestations. HLH is a rare and life-threatening syndrome characterized by uncontrolled immune activation and tissue destruction. The diagnostic criteria for HLH include fever, splenomegaly, bicytopenia, hypertriglyceridemia with or without hypofibrinogenemia, haemophagocytosis in bone marrow, spleen, lymph nodes or cerebrospinal fluid, reduced natural killer (NK) cell activity, ferritin >500 ng/mL, and soluble CD25 >2400 U/mL. Patients must fulfil at least 5/8 criteria to reach a diagnosis of HLH (Henter et al. 2007). Notably, some patients may present with atypical features, including neurological symptoms (ataxia, seizures).
Herein, we report the clinical course of a young boy who first presented at age 15 months with fever, bicytopenia, encephalopathy, and seizures. Subsequent whole exome sequencing (WES) identified genetic aberrations in the LRBA gene, encoding LRBA.
Methods
Chart review
Patient data was gathered by comprehensive retrospective chart review, including clinical documentation, immunological investigations, and targeted gene sequencing results. Informed consent was obtained from the family in accordance with the SickKids Research Ethics Board.
Genetic analysis
DNA from blood was submitted to The Centre for Applied Genomics (TCAG), Toronto, Canada, for exome library preparation and sequencing. DNA was quantified by Qubit DNA HS assay (Life Technologies, Carlsbad, CA) and 100 ng of input DNA was used for library preparation using the Ion AmpliSeq Exome Kit (Life Technologies) according to the manufacturer’s recommendations. The Ampliseq Exome library was immobilized on Ion PI™ Ion Sphere™ particles using the Ion PI Template OT2 200 Kit v3. Sequencing was performed with the Ion PI Sequencing 200 Kit v3 and Ion PI Chip v2 in the Ion Proton™ semiconductor sequencing system following the manufacturer’s recommendation. Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server, using the Ion Proton ampliseq germline low stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline based on Annovar (November 2014 version) and RefSeq gene models (downloaded from UCSC 01 August 2015).
Results
Clinical features
Our male patient was born at term with no significant antenatal and postnatal complications. He was well during his first year of life, and thriving with normal development. At 15 months of age, he presented with prodrome of fever and reduced appetite for 3 days and was managed symptomatically at home. On day 3 of illness, he developed acute onset flaccid tone with eye deviation to the right and jerking movement of the distal limbs, which was treated with midazolam. He was subsequently admitted to the local hospital for febrile seizure. Four days later, he had seizure recurrence with a generalized tonic-clonic seizure with eye deviation to the left, which was sustained for 30 minutes. He had significantly decreased level of consciousness after the event and was encephalopathic.
His parents are both of Pakistani descent and consanguineous (first cousins), and he has 3 healthy older sisters. There is no significant family history of recurrent infection, metabolic, or genetic disease. His mother is healthy and father reports underlying congenital absence of vas deferens.
Our patient was referred to SickKids Hospital for further evaluation and management and subsequently admitted to ICU. Clinical examination revealed a thriving child with no dysmorphic features. There was no lymphadenopathy, and cardiovascular and respiratory examination was normal. Abdominal examination revealed an enlarged liver 5 cm below the costal margin, however, there was no splenomegaly. Neurological examination revealed abnormal posturing, increased tone, and brisk reflexes.
Laboratory evaluation
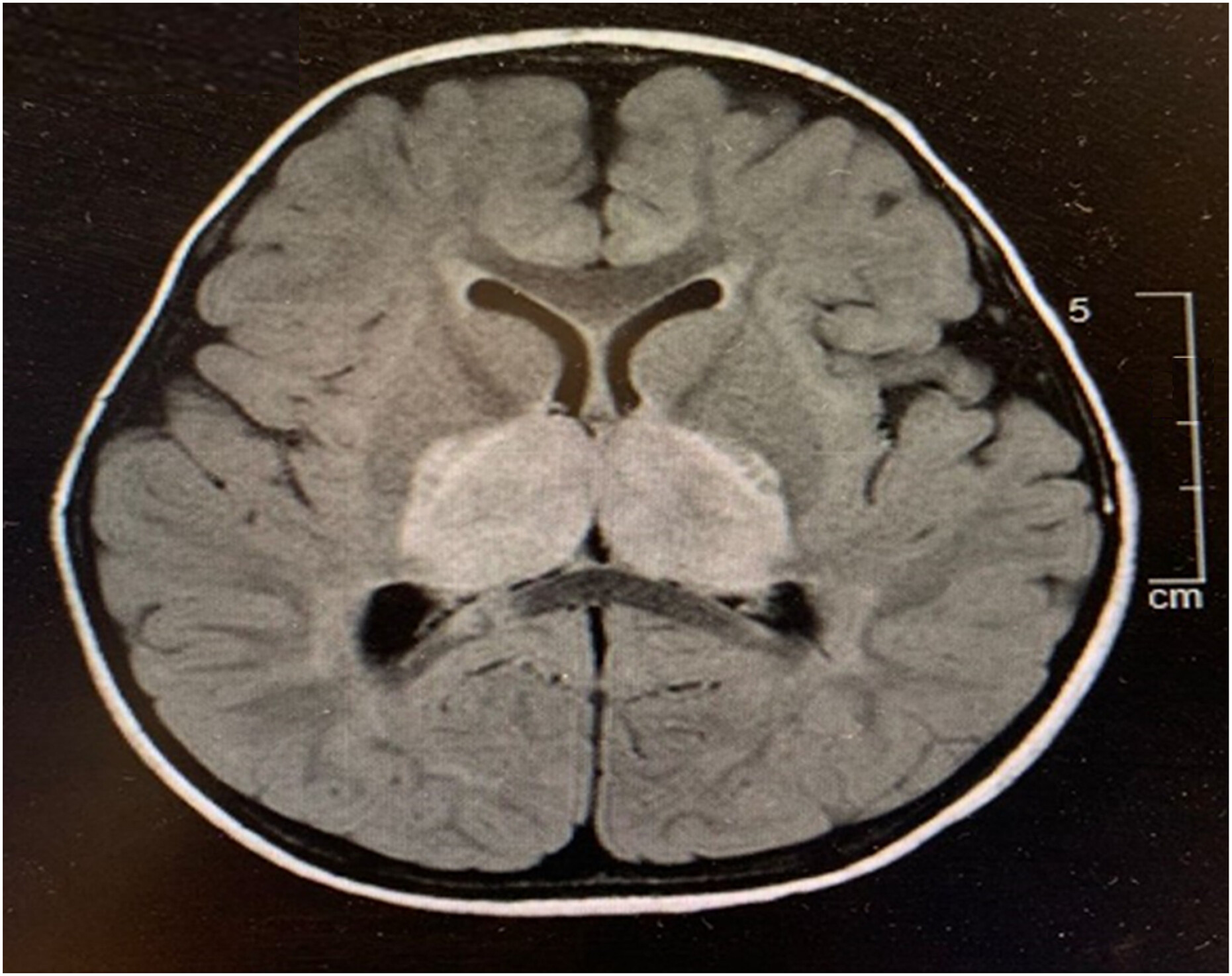
Laboratory evaluations (Table 1) revealed leukopenia and anaemia with thrombocytosis. His absolute neutrophil count was low. Liver enzymes alanine transaminase (ALT), aspartate aminotransferase (AST) and gamma-glutamyl transferase (GGT) were elevated. Nasopharyngeal swab for viruses were negative, including for Influenza. Other infectious work-up which included serology for Bartonella, West Nile Virus/Flavivirus, Dengue, Parvo, Lyme, syphilis, Mycoplasma, cytomegalovirus (CMV) and Epstein-Barr virus (EBV) were all negative except for human herpesvirus 6 (HHV6). Lumbar puncture results revealed white blood cells (WBC) at 1 × 106/L, red blood cells (RBC) at 2 × 106/L, protein at 1.39 g/L, and glucose at 3.9 mmol/L. Cerebral spinal fluid (CSF)cultures were negative. CSF PCR was negative for herpes simplex virus (HSV), varicella zoster virus (VZV) and Mycoplasma, but positive for HHV6. He was evaluated for HLH and had elevated ferritin and triglyceride and low fibrinogen, however, he did not meet the diagnostic criteria for HLH. His immunoglobulin levels were normal. Bone marrow examination was inconclusive. Natural killer (NK) cell degranulation and CD56/perforin were within normal range. Soluble CD25 and CD163 were normal. Electroencephalogram and magnetic reasonance imaging (MRI) results (Figure 1, Figure 2) were suggestive of acute necrotizing encephalopathy. He received high dose pulse IV methylprednisolone (30 mg/kg for 5 days) and plasmapheresis for 4-daily treatments. He responded to treatment with marked neurological improvement and upon discharge had minimal neurological deficit. He continued to show improvement in his neurological parameters during subsequent follow-up.
Table 1:

Figure 1:

Figure 2:
Progression of illness
At 36 months of age, our patient presented to a local hospital in Pakistan with similar symptoms. He developed high grade fever, lethargy, and declining loss of consciousness. He subsequently developed severe encephalopathy, abnormal movements, and fixed dilated pupils. Blood investigations revealed elevated neutrophils, lactate, liver enzymes (ALT was 678 U/L), C-reactive protein, ferritin, and prolonged prothrombin time. Dengue serology was positive. Urgent computerized tomography (CT) scan of the brain was reported locally as having cerebral oedema and bilateral thalamic disease. He was unstable during his admission to the intensive care unit and lumbar puncture and MRI was contraindicated. He received IV immunoglobulins and high dose methylprednisolone therapy. Despite treatment, he succumbed to his illness.
Genetic testing
Initial genetic testing included targeted sequencing of the RANBP2 gene, which was returned negative. An autoinflammatory panel noted a variant of uncertain significance in the MEFV gene, however, this did not fit the clinical picture of our patient.
In the year following his death, the parents were re-referred to our complex immunology clinic for further evaluation of a possible primary immunodeficiency.
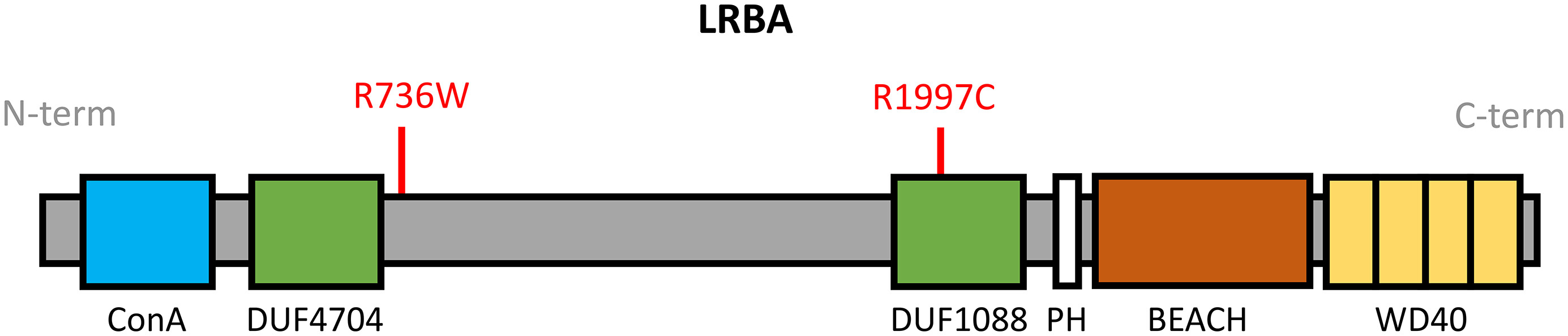
Research WES subsequently revealed a set of compound heterozygous missense mutations in the LRBA gene (NM_001364905). The results were confirmed in a clinical laboratory (Primary Immunodeficiency Panel, Invitae). The first variant, c.2206A>T, targets exon 18 resulting in the substitution of arginine with threonine, p.R736W (Figure 3), and has not been previously reported. It has an allele frequency of 0.0003099 in South Asians and 0 in other populations (gnomAD), and is predicted damaging by MutationAssessor (1.94). The second variant, c.5989C>T, targets exon 38 leading to substitution of cystine with threonine, p.R1997C (Figure 3). It is predicted damaging according to SIFT (0.004), PP2 (0.966), and MutationAssessor (2.28), and has an allele frequency of 0.03778 (gnomAD).
Figure 3:
Segregation analysis revealed the presence of the c.2206A>T (p.R736W) LRBA variant in the patient’s father, and the c.5989C>T (p.R1997C) variant in the patient’s mother.
Discussion
We describe here a patient with a set of compound heterozygous mutations in LRBA with atypical presentation of recurrent HLH including CNS manifestations, who was initially diagnosed with acute necrotizing encephalopathy of childhood (ANEC). Our patient had features of ANEC (from EEG and MRI) and responded to initial treatment with IVIG and plasmapheresis with good neurological outcome. He was suspected to have an underlying monogenic mutation based on family history of consanguinity. Recurrent ANEC secondary to underlying RANBP2 variants has previously been reported in patients with a positive family history or history of consanguinity (Suri 2010) (Neilson et al. 2009) (Bashiri et al. 2020). The negative findings on RANBP2 genetic sequencing prompted further genetic evaluation.
Research-based WES was essential for identifying the LRBA genetic aberrations in our patient and the interpretation will be an important consideration during future genetic counselling of his family members. Of the two variants described in this case study, c.2206A>T (p.R736W) is novel and previously unreported. The arginine residue at position 736 is perfectly conserved in vertebrates, moreover, the local region is highly conserved and in silico tools predict a functional impact. Regarding c.5989C>T (p.R1997C), the arginine residue at position 1997 is almost perfectly conserved in mammalians and vertebrates. The substitution with cysteine results in loss of a positive charge and marked shortening of the side chain. Thus, while the second mutation alone may not result in a significant clinical phenotype (as reflected by the higher frequency), when present in tandem, the combined impact of this set of compound heterozygous missense variants results in loss-of-function of LRBA. Our patient presented with an atypical phenotype of recurrent HLH with seizures and encephalopathy.
LRBA is a 2863 amino acid protein and contains multiple domains, including the Concanavalin A-like (ConA-like) domain, 2 domains of unknown function (DUF4704 and DUF1088), pleckstrin homology (PH) domain, beige and Chediak-Higashi (BEACH) domain, and WD40 repeat domains. The ConA-like domain located at the N-terminus binds to oligosaccharides and is involved in protein trafficking and sorting. The PH and BEACH domains form prominent grooves to recruit binding partners. Both the BEACH and WD40 repeat domains are involved in vesicle binding (Figure 3).
Biallelic LRBA loss-of-function is one of the most common causes of common variable immunodeficiency. Deficiency of LRBA and thus diminished expression of the inhibitory checkpoint molecule CTLA4 on the surface of T reg cells leads to unrestrained immune responses and autoimmune features. Notably, given the linked pathways of CTLA4 and LRBA, there is significant clinical overlap between LRBA deficiency and CTLA4 haploinsufficiency. Patients harboring compound heterozygous mutations in LRBA may present with a broader spectrum of manifestations, including autoimmune disease affecting multiple organs, chronic diarrhea, and organomegalies. Yao et al. describe a series of 5 cases with similar compound heterozygosity affecting LRBA (Yao et al. 2022). Interestingly, one of the cases was initially diagnosed with HLH due to fever, splenomegaly, elevated ferritin, low fibrinogen, hemophagocytosis, and hemopenia. He was successfully treated with HLH first-line chemotherapy. HLH-associated with a single heterozygous LRBA variant has also been reported (Ren et al. 2021) (Mo et al. 2019). Our patient was worked up for HLH given his fever and bicytopenia, however, while ferritin and triglyceride levels were elevated and fibrinogen was low, other investigations were within normal range and thus he did not fulfil HLH criteria. Further, his ferritin and triglyceride subsequently returned to normal. During his second bout of illness, prior to his demise, he had elevated liver enzymes and ferritin. We postulate that our patient had recurrent HLH, precipitated by 2 viral infections - HHV6 and dengue.
Neurological manifestations including seizures, altered consciousness, facial palsy, or difficulty in swallowing/speech have previously been reported in patients with HLH, affecting up to 46% of cases (Kim et al. 2012). In a large international cohort of 212 patients with LRBA deficiency, 12.3% were reported to have CNS effects, including seizure, neurodevelopment delay and demyelinating disorders (Jamee et al. 2021). Further, a report of Iranian cohorts identified neurological manifestations in 23% of the patients (Alkhairy et al. 2016), while Gamez-Diaz et al. reported that 5% of their patients with underlying LRBA deficiency presented with stroke (Gamez-Diaz et al. 2016).
Unfortunately, due to his atypical presentation and rigidity of the HLH criteria, our patient remained without a diagnosis until comprehensive next generation sequencing in the form of WES was performed. Similarly, we recently reported the diagnostic odyssey of a patient with early signs of HLH and whom did not satisfy the criteria for many years (Sham et al. 2022). It remains important for clinicians to consider atypical presentations of HLH as patients would benefit from specific medical therapy. In the case of LRBA deficiency, abatacept (a CTLA4-based fusion protein) has been shown to improve patient overall outcomes (Kiykim et al. 2019).
In summary, this case helps to expand the known spectrum of disease associated with compound heterozygous mutations in LRBA. WES serves as a useful tool to diagnose and guide treatment in complex immunological diseases and thus should be considered in patients with atypical manifestations.
REFERENCES
Alkhairy O.K., Abolhassani H., Rezaei N., Fang M., Andersen K.K., Chavoshzadeh Z., Mohammadzadeh I., El-Rajab M.A., Massaad M., Chou J., Aghamohammadi A., Geha R.S., and Hammarström L. 2016. Spectrum of Phenotypes Associated with Mutations in LRBA. J. Clin. Immunol. 36: 33–45.
Bashiri F.A., Johani S.A., Hamad M.H., Kentab A.Y., Alwadei A.H., Hundallah K., Hasan H.H., Alshuaibi W., Jad L., Alrifai M.T., Hudairi A, Sheikh R.A., Alenizi A., Alharthi N.A., Abdelmagid T.A., Ba-Armah D., Salih M.A., and Tabarki B. Oct 2020. Acute Necrotizing Encephalopathy of Childhood: A Multicenter Experience. Front. Pediatr. 8: 526.
Cagdas D., Halaçlı S.O., Tan Ç., Lo B., Çetinkaya P.G., Esenboğa S., Karaatmaca B., Matthews H., Balcı-Hayta B., Arıkoğlu T., Ezgü F., Aladağ E., Saltık-Temizel İ.N., Demir H., Kuşkonmaz B., Okur V., Gümrük F., Göker H., Çetinkaya D., Boztuğ K., Lenardo M., Sanal Ö., and Tezcan İ. 2019. A Spectrum of Clinical Findings from ALPS to CVID: Several Novel LRBA Defects. J. Clin. Immunol. 39: 726–738.
Gámez-Díaz L., August D., Stepensky P., Revel-Vilk S., Seidel M.G., Noriko M., Morio T., Worth A.J.J., Blessing J., de Veerdonk F.V., Feuchtinger T., Kanariou M., Schmitt-Graeff A., Jung S., Seneviratne S., Burns S., Belohradsky B.H., Rezaei N., Bakhtiar S., Speckmann C., Jordan M., and Grimbacher B. (2016). The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J. Aller. Clin. Immunol. 137(1): 223–230.
Henter J.-I., Horne A., Aricó M., Egeler R.M., Filipovich A.H., Imashuku S., Ladisch S., McClain K., Webb D., Winiarski J., and Janka G. 2007. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr. Blood Cancer, 48(2): 124–131.
Jamee M., Hosseinzadeh S., Sharifinejad N., Zaki-Dizaji M., Matloubi M., Hasani M., Baris S., Alsabbagh M., Lo B., and Azizi G. 2021. Comprehensive comparison between 222 CTLA-4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review. Clin. Exp. Immunol. 205: 28–43.
Kim M.-M., Yum M-S., Choi H-W., Ko T-S., Im H.J., Seo J-J., and Koh K-N. 2012. Central nervous system (CNS) involvement is a critical prognostic factor for hemophagocytic lymphohistiocytosis. Korean J. Hematol. 47(4): 273–280.
Kiykim A., Ogulur I., Dursun E., Charbonnier L.M., Nain E., Cekic S., Dogruel D., Karaca N.E., Cogurlu M.T., Bilir O.A., Cansever M., Kapakli H., Baser D., Kasap N., Kutlug S., Altintas D.U., Al-Shaibi A., Agrebi N., Kara M., Guven A., Somer A., Aydogmus C., Ayaz N.A., Metin A., Aydogan M., Uncuoglu A., Patiroglu T., Yildiran A., Guner S.N., Keles S., Reisli I., Aksu G., Kutukculer N., Kilic S.S., Yilmaz M., Karakoc-Aydiner E., Lo B., Ozen A., Chatila T.A., and Baris S. 2019. Abatacept as a Long-Term Targeted Therapy for LRBA Deficiency. J Aller. Clin. Immunol. Pract. 7(8): 2790–2800.
Kostel Bal S., Haskologlu S., Serwas N.K., Islamoglu C, Aytekin C., Kendirli T., Kuloglu Z., Yavuz G., Dalgic B., Siklar Z., Kansu A., Ensari A., Boztug K., Dogu F., and Ikinciogullari A. 2017. Multiple Presentations of LRBA Deficiency: a Single-Center Experience. J. Clin. Immunol. 37(8): 790–800.
Lo B., Zhang K., Lu W., Zheng L., Zhang Q., Kanellopoulou C., Zhang Y., Liu Z., Fritz J.M., Marsh R., Husami A, Kissell D., Nortman S., Chaturvedi V., Haines H., Young L.R., Mo J., Filipovich A.H., Bleesing J.J., Mustillo P, Stephens M., Rueda C.M., Chougnet C.A., Hoebe K., McElwee J., Hughes J.D., Karakoc-Aydiner E., Matthews H.F., Price S., Su H.C., Rao V.K., Lenardo M.J., and Jordan M.B. 2015. Patients with LRBA deficiency showCTLA4 loss and immune dysregulationresponsive to abatacept therapy. Science, 349(6246): 436–440.
Lopez-Herrera G., Tampella G., Pan-Hammarström Q., Herholz P., Trujillo-Vargas C.M., Phadwal K., Simon A.K., Moutschen M., Etzioni A., Mory A., Srugo I., Melamed D., Hultenby K., Liu C., Baronio M., Vitali M., Philippet P., Dideberg V., Aghamohammadi A., Rezaei N., Enright V., Du L., Salzer U., Eibel H., Pfeifer D., Veelken H., Stauss H., Lougaris V., Plebani A., Gertz E.M., Schäffer A.A., Hammarström L., and Grimbacher B. 2012. Deleterious Mutations in LRBA Are Associated with a Syndrome of Immune Deficiency and Autoimmunity. Am. J. Human Gen. 90: 986–1001.
Mo W., Wei W., Sun Y., Yang Y., Guan Z., Li M., Zhu P., and Chi Z. 2019. Application of blood and immunodeficiency gene detection in the diagnosis of hemophagocytic lymphohistiocytosis patients. Exper. Hematol. 78: 62–69.
Neilson D.E., Adams M.D., Orr C.M.D., Schelling D.K., Eiben R.M., Kerr D.S., Anderson J., Bassuk A.G., Bye A.M., Childs A-M., Clarke A., Crow Y.J., Rocco M.D., Dohna-Schwake C., Dueckers G., Fasano A.E., Gika A.D., Gionnis D., Gorman M.P., Grattan-Smith P.J., Hackenberg A., Kuster A., Lentschig M.G., Lopez-Laso E., Marco E.J., Mastroyianni S., Perrier J., Schmitt-Mechelke T., Servidei S., Skardoutsou A., Uldall P., der Knaap M.S.V., Goglin K.C., Tefft D.L., Aubin C., de Jager P., Hafler D., and Warman M.L. 2009. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am. J. Hum. Genet. 84(1): 44–51.
Ren Y., Xiao F., Cheng F., Huang X., Li J., Wang X., Lang W., Zhou X., Lan J., and Tong H. (2021). Whole exome sequencing reveals a novel LRBA mutation and clonal hematopoiesis in a common variable immunodeficiency patient presented with hemophagocytic lymphohistiocytosis. Exper. Hematol. Oncol. 10(1): 38.
Sham M., Zhu R., and Pasternak Y. 2022. Whole exome sequencing identifies causative compound heterozygous variants in PRF1 in late-onset familial hemophagocytic lymphohistiocytosis. LymphoSign J. 9(4).
Suri M. 2010. Genetic basis for acute necrotizing encephalopathy of childhood. Dev. Med. Child Neurol. 52(1) 4–5.
Yao J., Gu H., Mou W., Chen Z., Ma J., Ma H., Li N., Zhang R., Wang T., Jiang J., and Wu R. 2022. Various phenotypes of LRBA gene with compound heterozygous variation: A case series report of pediatric cytopenia patients. International Journal of Immunopathology and Pharmacology 36: 3946320221125591.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 10 • Number 1 • March 2023
Pages: 20 - 26
History
Received: 24 March 2023
Accepted: 30 March 2023
Accepted manuscript online: 30 March 2023
Copyright
© 2023.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
MarinaSham, DanieleMerico, MyraPereira, Chaim M.Roifman, and MaianRoifman. 2023. Identification of novel compound heterozygous LRBA mutations associated with recurrent hemophagocytic lymphohistiocytosis and CNS manifestations. LymphoSign Journal.
10(1): 20-26. https://doi.org/10.14785/lymphosign-2023-0004
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
