Identification of a novel NFKB2 mutation in a patient presenting with autoimmune cytopenia and generalized granulomatous lymphadenopathy
Abstract
Introduction: NF-κB proteins are transcription factors that modulate various functions of the immune system. NF-κB2 (or p100/p52) has particularly important roles in B cell development and function. Primary immunodeficiency due to mutations in the NFKB2 gene, encoding NF-κB2, range from combined immunodeficiency with susceptibility to viral or opportunistic infections to primarily antibody deficiency.
Methods: A comprehensive chart review of our patient was performed.
Results: Our patient, currently a 19-year-old male, presented with multiple autoimmune cytopenia resistant to treatment and generalized granulomatous lymphadenopathy. Whole exome sequencing identified a novel pathogenic variant in NFKB2 (c.1700C>T; p.A567V) that is the cause of our patient’s presentation.
Conclusion: We present a novel pathogenic variant in NFKB2 with an unusual presentation.
Statement of novelty: Here, we report a novel mutation in NFKB2 and the clinical presentation of the affected patient, which helps in further understanding the NF-κB2 pathway and its associated disease.
Introduction
Common Variable Immunodeficiency (CVID) is a heterogenous clinical diagnosis characterized by hypogammaglobinemia and poor antibody response to specific antigens, leading to an increased susceptibility to infections. It is the most common symptomatic primary immunodeficiency, with an estimated prevalence of 1:10 000 to 1:100 000 depending on race and geographical region. While it is a clinical diagnosis, monogenic causes of CVID have been identified in up to 10% of CVID cases (Szczawinska-Poplonyk et al. 2021; Bogaert et al. 2016). One such monogenic cause is NFKB2 pathogenic variants.
Nuclear factor kappa B (NF-κB) proteins are transcription factors that reside in the cytoplasm until activated, upon which they migrate to the nucleus and bind to their gene targets. The NF-κB signal transduction pathway has important roles in inflammation and host immune responses (Hayden and Ghosh 2011). While NF-κB1 has broad immune functions, NF-κB2, a key part of the non-canonical NF-κB pathway, is more specialized to B cell development and maturation. The non-canonical pathway also has functions in secondary lymphoid organogenesis, thymic epithelial differentiation, and bone metabolism (Sun 2012; Hayden and Ghosh 2011).
NF-κB2 (also known as p100/p52) is encoded by the NFKB2 gene. Upon activation of its pathway by signals from cluster of differentiation 40 ligand (CD40L), lymphotoxin β receptor (LTβR), and B cell-activating factor receptor (BAFFR), NF-κB-inducing kinase (NIK) is activated, phosphorylating the homodimer IKKα/IKKα, which transfers the phosphate group to the C-terminal residues of p100. Subsequently, p100 is converted by proteasomal processing of its C-terminal into p52, the mature form of the NF-κB2 transcription factor. The p52 subunit can dimerize with other members of the NF-κB family, however, preferentially makes RelB/p52 dimers which are translocated to the nucleus where they regulate target gene expression (Pires et al. 2018; Sun 2012).
Immunodeficiency due to a heterozygous mutation in NF-κB2 was first described in 2013 as an early onset CVID associated with central adrenal insufficiency (adrenocorticotropin “ACTH” deficiency) (Chen et al. 2013), the so-called DAVID syndrome (deficit in anterior pituitary function and variable immune deficiency) (Quentien et al. 2012). Since then, other cases have been reported with and without anterior pituitary defects and with varying immune effects including CVID, autoimmunity, and combined immunodeficiency.
Here, we report the case of a patient who presented with autoimmune cytopenia and generalized granulomatous lymphadenopathy. Whole exome sequencing subsequently identified a novel causative missense variant in the NFKB2 gene.
Methods
Patient information
Data were compiled prospectively and retrospectively from medical records in accordance with research ethics approved protocols.
Serum concentration of immunoglobulins and specific antibodies
Nephelometry was used to determine serum concentrations of immunoglobulins (IgG, IgA, and IgM) Serum antibody levels to tetanus toxoid and pneumococcus (pneumococcal capsular polysaccharide) were quantified by enzyme-linked immunosorbent assay (ELISA), as per the manufacturer’s instructions (Binding Site, Birmingham, UK). Serum antibodies to measles, mumps, and rubella were also measured by ELISA (Euroimmune kits, Gross-Groenau, Germany). Serum isohemagglutinin titers are reported as the dilution at which macroscopic agglutination is observed (antiglobulin phase) and was determined by twofold serial dilution with erythrocytes.
T cell proliferative responses
Lymphocyte proliferative responses to mitogens, including PHA, anti-CD3 and anti-CD28 antibodies, as well as a panel of recall antigens (Candida, Tetanus toxoid, Herpes zoster, and CMV), was determined by thymidine incorporation, as reported previously. All assays were performed in triplicate and were compared with random normal controls.
Exome sequencing and variant calling
DNA from blood was submitted to The Centre for Applied Genomics (TCAG), Toronto, Canada for exome library preparation and sequencing. DNA was quantified by Qubit DNA HS assay (Life Technologies, Carlsbad, CA) and 100 ng of input DNA was used for library preparation using the Ion AmpliSeq Exome Kit (Life Technologies) according to the manufacturer’s recommendations. The Ampliseq Exome library was immobilized on Ion PI™ Ion Sphere™ particles using the Ion PI Template OT2 200 Kit v3. Sequencing was performed with the Ion PI Sequencing 200 Kit v3 and Ion PI Chip v2 in the Ion Proton™ semiconductor sequencing system following the manufacturer’s recommendation. Alignment and variant calling were performed using Torrent Suite (v4.0) on the Ion Proton Server, using the Ion Proton ampliseq germline low stringency setting and the hg19 reference genome. The variants were annotated using an in-house annotation pipeline based on Annovar (November 2014 version) (Wang et al. 2010) and RefSeq gene models (downloaded from UCSC 01 August 2015).
Sequencing analysis
The patient’s genomic DNA was extracted from peripheral blood lymphocytes using the Geneaid Genomic DNA Mini Kit. Genomic DNA was amplified by PCR with specific primers designed upstream and downstream of the MSN gene. Sequencing was done using GenomeLab Dye Terminator Cycle Sequencing (DTCS) Quick Start Kit (Beckman Coulter) and analyzed on CEQ 8000 Genetic Analysis System (Beckman Coulter).
Case presentation
Our patient is currently a 19-year-old male who first presented at the age of 2 with thrombocytopenia (Table 1), splenomegaly, and generalized lymphadenopathy. He was initially given the diagnosis of immune thrombocytopenia (ITP) and was treated with intravenous immunoglobulin replacement therapy (IVIG). Over the following year, he required 4 doses of IVIG and was thus diagnosed with chronic ITP. At the age of 3, he was referred to our Immunology clinic for assessment due to his persistent ITP and generalized lymphadenopathy. Other than several viral upper respiratory tract infections, he had no history of recurrent or severe infections. Immune evaluation revealed normal total immunoglobulin levels (Table 2), but given his recent IVIG, further humoral assessment was difficult. CD4+, CD8+ and CD19+ cell counts at the time were normal and a decrease in his total CD16+56+ cells was noted (Table 3). His lymphocyte proliferation responses to the mitogen phytohemagglutinin (PHA) were within normal range, although antigen recall responses (day 6) were low (<20 stimulation index) against Candida and Herpes zoster (Table 4). Screening for ALPS with double negative T cells was negative. He also underwent an assessment by Rheumatology at the same time and their work up was not suggestive of a rheumatologic disease.
Table 1:
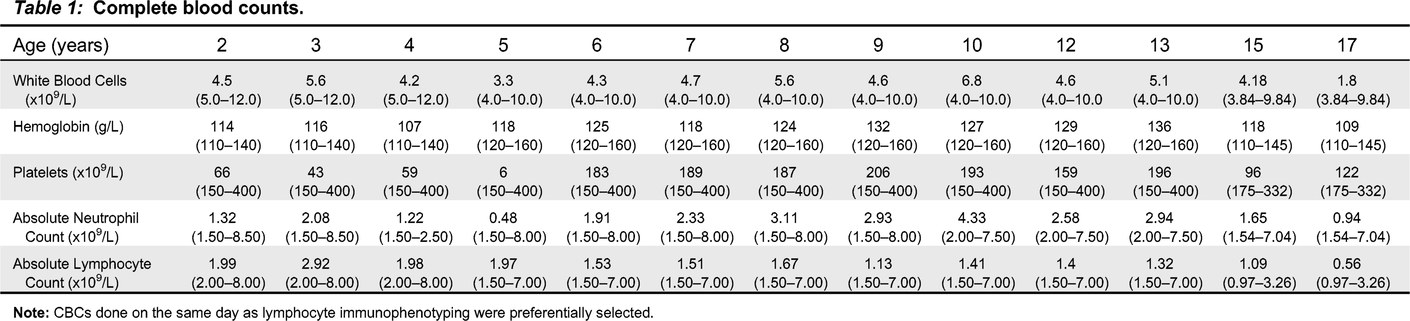
Note: CBCs done on the same day as lymphocyte immunophenotyping were preferentially selected.
Table 2:

Note: Ab: Antibody, IgG: Immunoglobulin G, IgA: Immunoglobulin A, IgM: Immunoglobulin M, pre: before vaccination or booster, post: after vaccination or booster.
Table 3:

Table 4:
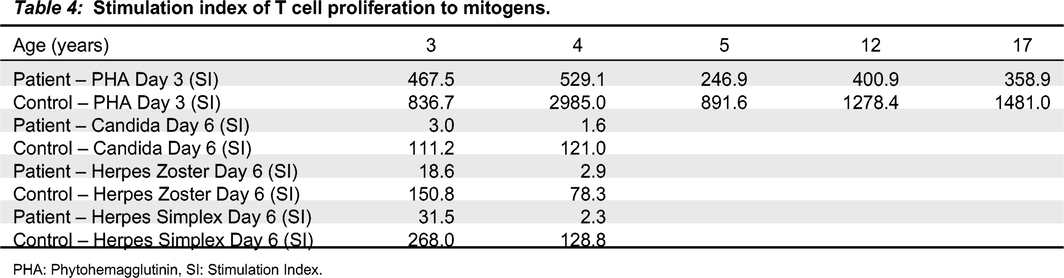
PHA: Phytohemagglutinin, SI: Stimulation Index.
In the following months, he continued to have recurring thrombocytopenia that was treated with anti-D and steroids. A reassessment of his humoral immunity was done 6 months post-IVIG and was normal.
At the age of 5, he developed autoimmune hemolytic anemia. He was known to be direct coomb’s positive since his initial presentation and was treated with oral steroids at the time. He continued to have recurrent thrombocytopenia and developed neutropenia which was thought to be autoimmune as well. A bone marrow biopsy at the time was normal and reactive.
One year later, at the age of 6, he had several episodes of ITP in a short duration, all treated with IVIG. Given the severity and persistence of his ITP, it was decided to start him on mycophenolate mofetil (MMF). A repeat bone marrow biopsy prior to the initiation of MMF was normal. After starting MMF, his cytopenia resolved and he had no recurrences of thrombocytopenia or hemolytic anemia.
After the initiation of MMF, he had a progressive decrease in his immunoglobulins and loss of specific antibodies to vaccine preventable infections (Table 2). He did not have any recurrent sinopulmonary infections. It was unclear at the time whether the hypogammaglobinemia was due to MMF treatment, GI losses, his underlying disease, or a combination of the above. Due to the persistent and significant decrease in his antibody levels he was started on regular IVIG at the age of 14, initially every 4 weeks then every 3 weeks due to persistently low IgG. He was transitioned to subcutaneous immunoglobulins at the age of 18.
Over the years he has developed multiple other complications summarized below:
Varicella infection
At the age of 8, he had a contact at school who developed chicken pox. He was given VZIG as post-exposure prophylaxis but still developed a fever, typical varicella rash, and hip pain about 2 weeks later. He was admitted to hospital and received a 4-day course of intravenous acyclovir. He was then discharged in a good condition and received a further 10 days of oral acyclovir. He had no complications of this infection.
Lymphadenopathy with granulomatous disease
Since presentation, he showed wide-spread lymphadenopathy, including neck, groin, chest and abdominal (mesenteric and retroperitoneal) adenopathy. He had biopsies of his lymph nodes on multiple occasions, most recently an inguinal lymph node at age 16 and an abdominal lymph node at age 17, both showing reactive lymphoid hyperplasia with non-necrotizing granulomas. Notably, he has never shown evidence of hypercalcemia or elevated vitamin D. His most recent abdominal ultrasound showed stable mild hepatomegaly, moderate splenomegaly, and stable mesenteric and iliac lymphadenopathy.
Hepatosplenomegaly with chronic mild elevation of liver enzymes
The patient has had a long-standing history of hepatosplenomegaly, with mild elevation of his liver enzymes. He did intermittently report on clay-colored stools (once every few weeks) but never had consistent concern for acholic stools. This was followed by gastroenterology, with no intervention, and has been stable for several years.
Duodenitis and protein losing enteropathy
From the age of 9, he began complaining of recurrent abdominal pain and bloating. He was also concurrently showing poor weight gain. This was initially attributed to mesenteric adenopathy, but on endoscopy he was found to have duodenitis with intraepithelial lymphocytosis and villous blunting. A diagnosis of celiac disease was ruled out, and it was suspected that this could be iatrogenic secondary to MMF or a manifestation of his underlying disease. He went on to develop chronic loose stools and protein-losing enteropathy, but since the age of 15, his stools have stabilized, and at present he reports only 1 bowel movement per day, although still loose. His serum albumin has also stabilized. He is not currently receiving any active intervention from the GI team.
Delayed puberty and short stature
He had an endocrinology assessment due to these concerns with no identified diagnosis beyond delayed bone age on initial assessments. As part of a work-up to rule out hypopituitarism he had a brain MRI which showed a pars intermedia cyst, but this is not believed to be of clinical significance. Evaluation of his Hypothalamic-Pituitary-Adrenal (HPA) axis has been within normal limits. Over the past few years, he has made gains in both height and weight and has not required active intervention from endocrinology.
Bilateral nephrocalcinosis
On a previous ultrasound to monitor his organomegaly and lymphadenopathy, he was incidentally found to have bilateral nephrocalcinosis, which has always been asymptomatic. He has had extensive work-up by nephrology revealing only low urine citrate of unknown etiology. To date, he is treated with citrate supplementation.
Bilateral lung parenchyma interstitial thickening
At the age of 16, as part of a work-up for his lymphadenopathy, our patient had a chest CT showing bilateral diffuse lung parenchyma interstitial thickening and nodular opacities. He was entirely asymptomatic at the time. He had a broncho-alveolar lavage which showed lymphocytosis (37%–39%). An extensive infectious work-up was negative, and pulmonary function tests showed normal volumes. He has never reported any symptoms related to these findings.
Genetic evaluation
Genetic testing was performed during his initial presentation for genes associated with ALPS with negative results. He later underwent research-based whole exome sequencing which identified a heterozygous variant of unknown significance in NFKB2 (NM_001077494), c.1700C>T (p. A567V). This was verified by Sanger sequencing in a clinical laboratory. On segregation studies, it was found that his mother is a healthy carrier of the same variant.
On his most recent follow up, our patient reported that he is doing quite well on MMF and SCIG with no significant infections, no GI symptoms, and good weight gain.
Discussion
Here we describe a novel pathogenic variant in NFKB2 in a patient who presented with persistent autoimmune thrombocytopenia, hemolytic anemia, and neutropenia, as well as granulomatous generalized lymphadenopathy, and splenomegaly. The patient also developed hypogammaglobinemia by the age of 6 years, although he was on immunosuppressive therapy at the time with MMF. Other clinical manifestations as noted above included protein losing enteropathy, delayed puberty, short stature, bilateral nephrocalcinosis, and asymptomatic interstitial lung infiltrates.
The variant we report at c.1700C>T is a missense variant targeting the ankyrin rich domain of p100, the precursor protein that is encoded by the NFKB2 gene (see Figure 1). It changes the amino acid sequence from alanine to valine at position 567. The variant is predicted by CADD and SIFT to be damaging. The cytosine residue at the c.1700 locus is also highly conserved in all vertebrate species. This variant has an allele frequency of 2.87 × 10−5 in gnomAD, and has been reported in ClinVar once as a variant of unknown significance.
Figure 1:
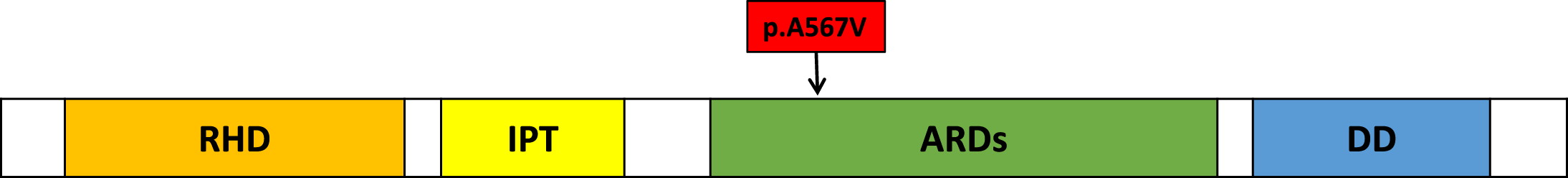
In contrast to most patients with NFKB2 mutations, our patient presented initially with autoimmune cytopenia and generalized lymphadenopathy. While this presentation has been reported in other patients, most patients with NFKB2 mutations initially present with recurrent infections and variable immunodeficiency (Klemann et al. 2019). Four other patients with pathogenic variants in the same domain as our patient were found in the literature but their presentations were not similar (Klemann et al. 2019; Kuehn et al. 2017).
Although our patient’s hypogammaglobinemia did not develop until after starting MMF and the onset of his gastrointestinal complaints, he did show early signs of antibody deficiency with early loss of antibodies to pneumococcus post-vaccination and low isohemagglutinin titers prior to MMF initiation. The immunosuppressive therapy may have accelerated the progression of his antibody deficiency but is unlikely to be the sole cause of it.
Lymphadenopathy has been reported in multiple patients (Klemann et al. 2019) and granulomatous disease has been reported in some patients as well (Kuehn et al. 2017; Brue et al. 2014). Enteropathy and lymphocytic lung infiltrations are also known possible manifestations of this disease. (Klemann et al. 2019).
Many patients with NFKB2 mutations have shown T cell mediated autoimmunity in the form of alopecia and (or) trachyonychia which did not manifest in our patient. (Pérez Cavazos et al. 2021; Bienias et al. 2021; Klemann et al. 2019). Our patient also did not show any ACTH or other anterior pituitary hormonal deficiency which has been reported in other patients (Klemann et al. 2019) despite his delayed puberty which was likely constitutional.
Some patients have been reported with severe viral or opportunistic infections, lymphopenia, and a combined immune defect (Abraham et al. 2021; Pérez Cavazos et al. 2021; Bienias et al. 2021; Klemann et al. 2019). We also did not observe this in our patient, however, he did have a mild decrease in his T cell proliferation to mitogens compared to control on several occasions.
In summary, we present a novel pathogenic variant in NFKB2 in a patient who presented primarily with autoimmune cytopenia and generalized granulomatous lymphadenopathy.
REFERENCES
Abraham R.S., Marshall J.M., Kuehn H.S., Rueda C.M., Gibbs A., Guider W., Stewart C., Rosenzweig S.D., Wang H., Jean S., and Peeples M. 2021. Severe SARS-CoV-2 disease in the context of a NF-κB2 loss-of-function pathogenic variant. J. Allergy Clin. Immunol. 147(2): 532–544.
Bienias M., Gabrielyan A., Geberzahn L., Rösen-Wolff A., Huebner A., Jacobsen E.M., Toepfner N., Fang M., Lee-Kirsch M.A., Roesler J., and Schuetz C. 2021. More severe than CVID: Combined immunodeficiency due to a novel NFKB2 mutation. Pediatr. Allergy Immunol. 32(4): 793–797.
Bogaert D.J., Dullaers M., Lambrecht B.N., Vermaelen K.Y., De Baere E., and Haerynck F. 2016. Genes associated with common variable immunodeficiency: One diagnosis to rule them all?. J. Med. Genet. 53(9): 575–590.
Brue T., Quentien M.H., Khetchoumian K., Bensa M., Capo-Chichi J.M., Delemer B., Balsalobre A., Nassif C., Papadimitriou D.T., Pagnier A., and Hasselmann C. 2014. Mutations in NFKB2and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med. Genet. 15(1): 1–7.
Chen K., Coonrod E.M., Kumánovics A., Franks Z.F., Durtschi J.D., Margraf R.L., Wu W., Heikal N.M., Augustine N.H., Ridge P.G., and Hill H.R. 2013. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 93(5): 812–824.
Hayden M.S. and Ghosh S. 2011. NF-κB in immunobiology. Cell Res. 21(2): 223–244.
Klemann C., Camacho-Ordonez N., Yang L., Eskandarian Z., Rojas-Restrepo J.L., Frede N., Bulashevska A., Heeg M., Al-Ddafari M.S., Premm J., and Seidl M. 2019. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front. Immunol. 10: 297.
Kuehn H.S., Niemela J.E., Sreedhara K., Stoddard J.L., Grossman J., Wysocki C.A., De La Morena M.T., Garofalo M., Inlora J., Snyder M.P., and Lewis D.B. 2017. Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood, 130(13): 1553–1564.
Pérez Cavazos S., De la Cruz Cruz R.A., Castillo Bejarano J.I., Vaquera Aparicio D.N., Mascarenas de Los Santos A.H., and Zárate Hernández M.D.C. 2021. Disseminated coccidioidomycosis as the first presentation of a C-Terminal NFKB2 pathogenic variant: A case report and review of the literature. Pediatr. Infect. Dis. J. 41(2): 140–144.
Pires B.R., Silva R.C., Ferreira G.M., and Abdelhay E. 2018. NF-kappaB: two sides of the same coin. Genes (Basel), 9(1): 24.
Quentien M.H., Delemer B., Papadimitriou D.T., Souchon P.F., Jaussaud R., Pagnier A., Munzer M., Jullien N., Reynaud R., Galon-Faure N., and Enjalbert A. 2012. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J. Clin. Endocrinol. Metab. 97(1): E121–E128.
Sun S.C. 2012. The noncanonical NF-κB pathway. Immunol. Rev. 246(1): 125–140.
Szczawinska-Poplonyk A., Schwartzmann E., Bukowska-Olech E., Biernat M., Gattner S., Korobacz T., Nowicki F., and Wiczuk-Wiczewska M. 2021. The pediatric common variable immunodeficiency—from genetics to therapy: A review. Eur. J. Pediatr. 181: 1371–1383.
Wang K., Li M., and Hakonarson H. 2010. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38(16): e164–e164.
Information & Authors
Information
Published In

LymphoSign Journal
Volume 10 • Number 1 • March 2023
Pages: 7 - 14
History
Received: 13 February 2023
Accepted: 2 March 2023
Accepted manuscript online: 8 March 2023
Version of record online: 20 April 2023
Copyright
© 2023.
Authors
Metrics & Citations
Metrics
Other Metrics
Citations
Cite As
AbdulrahmanAl Ghamdi, MarinaSham, LauraAbrego Fuentes, and LindaVong. 2023. Identification of a novel NFKB2 mutation in a patient presenting with autoimmune cytopenia and generalized granulomatous lymphadenopathy. LymphoSign Journal.
10(1): 7-14. https://doi.org/10.14785/lymphosign-2023-0001
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
There are no citations for this item
